- Record: found
- Abstract: found
- Article: found
Rett syndrome: a neurological disorder with metabolic components
Read this article at
Abstract
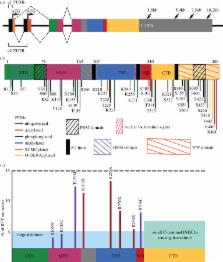
Rett syndrome (RTT) is a neurological disorder caused by mutations in the X-linked gene methyl-CpG-binding protein 2 ( MECP2), a ubiquitously expressed transcriptional regulator. Despite remarkable scientific progress since its discovery, the mechanism by which MECP2 mutations cause RTT symptoms is largely unknown. Consequently, treatment options for patients are currently limited and centred on symptom relief. Thought to be an entirely neurological disorder, RTT research has focused on the role of MECP2 in the central nervous system. However, the variety of phenotypes identified in Mecp2 mutant mouse models and RTT patients implicate important roles for MeCP2 in peripheral systems. Here, we review the history of RTT, highlighting breakthroughs in the field that have led us to present day. We explore the current evidence supporting metabolic dysfunction as a component of RTT, presenting recent studies that have revealed perturbed lipid metabolism in the brain and peripheral tissues of mouse models and patients. Such findings may have an impact on the quality of life of RTT patients as both dietary and drug intervention can alter lipid metabolism. Ultimately, we conclude that a thorough knowledge of MeCP2's varied functional targets in the brain and body will be required to treat this complex syndrome.
Related collections
Most cited references202
- Record: found
- Abstract: found
- Article: not found
Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2.
- Record: found
- Abstract: found
- Article: not found
A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome.
- Record: found
- Abstract: found
- Article: not found