- Record: found
- Abstract: found
- Article: found
The role of lysosomes in alpha-synucleinopathies: a focus on glial cells
other
10 December 2021
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Lysosomes are the major degradative compartments within eukaryotic cells. Besides
their role in the degradation and recycling of intra- and extracellular molecules,
they further mediate important biological processes, such as immune signaling and
perpetuation of nutrient- and energy homeostasis. Impairment of lysosomal function
triggers the accumulation of catabolic products within the organelle resulting in
lysosomal storage disorders (LSDs). Interestingly, clinical, molecular, and genetic
studies further indicate a strong link between lysosomal dysfunction and neurodegenerative
disorders, including Parkinson's disease (PD). Because of the association of lysosomal
dysfunction and protein aggregation of α-synuclein (α-Syn) in PD or multiple system
atrophy (MSA), the role of lysosomal pathways has been a matter of recent studies,
mostly focusing on neuronal cells. Although it is known that glial cells play an important
role in disease pathology of PD and MSA, only few studies on the lysosomal pathways,
within glial cells have been carried out. Hence, a better understanding of lysosomal
function in glia is needed to elucidate disease pathogenesis and to search for novel
therapeutic approaches.
Lysosomal biogenesis: The prerequisite for unhampered lysosomal degradation is constituted
by about 60 different acidic hydrolases and approximately 25 integral lysosomal membrane
proteins. The latter preserves the acidic pH within the lysosomal lumen, maintain
the ionic gradient and homeostasis, transport proteins into the lysosome, dispose
catabolism products into the cytosol, and are important for membrane trafficking/fusion.
The pathways in which lysosomes receive their biomaterial for the catabolic processing
involve three main types of autophagy: micro- and macroautophagy, as well as chaperone-mediated
autophagy (Trivedi et al., 2020). Lysosomal dysfunction, depending on the genetic
defect and biochemical property of the accumulating substrate, can lead to severe
pathology, accompanied with deficits in the central nervous system (CNS). There are
several therapeutic strategies for LSDs which focus on either increasing the activity
of the specific target enzyme, reducing substrate production, or modulating lysosomal
exocytosis (Kreher et al., 2021).
Role of lysosomes in neurons during synucleinopathies: An efficient lysosomal function
is pivotal for neuronal survival. As neurons reach a post-mitotic state after differentiation,
the degradation of neurotoxic protein aggregates is essential to prevent an irreversible
loss of neurons. Focusing on α-synucleinopathies, the aggregation of α-Syn represents
a key factor for the development of neurodegenerative disorders like PD or MSA. There
are several hypotheses for the occurrence of pathological α-Syn species and their
toxicity, especially within dopaminergic neurons in the substantia nigra. It is suggested
that in the early stages of PD, α-Syn pathology could have its origin in the olfactory
bulb, or the dorsal motor nucleus of the vagus. Recent studies have identified the
possibility of α-Syn aggregates to spread and seed pathology from cell-to-cell, reaching
dopaminergic neurons by distribution through synaptic coupled networks. Further, it
was observed that oxidative stress can trigger the conversion of the neurotransmitter
dopamine into a reactive quinone species within dopaminergic neurons, which might
contribute to their vulnerability. This reactive compound was shown to further cause
lysosomal dysfunction, disturb lysosomal enzyme activity, and cause neurotoxicity
(Surmeier, 2018).
Next to proteasomal processing, α-Syn can also get degraded via autophagy within neuronal
cells. Recent studies implicate the lysosomal proteases cathepsin D (CTSD), cathepsin
B (CTSB), and cathepsin L in the degradation of α-Syn. Additionally, impairments in
the lysosomal degradation pathway or enzyme dysfunction can lead to the aggregation
of α-Syn. Hence, disease-associated CTSD variants were shown to be impaired in their
maturation and exhibited altered α-Syn degradation properties in human cell models
(Bunk et al., 2021). Furthermore, it is known that mutations within the gene encoding
for the lysosomal enzyme β-glucocerebrosidase (GBA1) (
Figure 1
) present a common genetic risk factor for the development of PD. GBA1 gene mutations
in PD patients lead to an insufficient degradation of its substrate glucosylceramide
in lysosomes, which has been shown to interfere and accelerate α-Syn aggregation (Zunke
et al., 2018). Interestingly, the aggregation of α-Syn also contributes to further
lysosomal dysfunction, probably by interrupting lysosomal protein trafficking. Genetic
studies have associated further genes linked to lysosomal function, like the lysosomal
hydrolases CTSD and CTSB, the cation-transporting ATPase 13A2 (ATP13A2) or vacuolar
protein sorting-associated protein 35 (VPS35) to an increased risk of developing PD
(
Figure 1
), indicating the importance of lysosomal function in neuronal homeostasis (Puska
et al., 2018; Zunke et al., 2018).
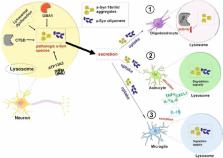
Figure 1
Effects of secreted neuronal α-synuclein (α-Syn) species on different glial cells
in the context of lysosomal pathways.
Lysosomal dysfunction as well as mutations within lysosomal enzymes, as for example
CTSD and GBA1, or lysosomal membrane transport protein ATP13A2 can trigger the formation
of pathologic α-Syn species (fibrils/aggregates, oligomers) in neurons and potentially
in lysosomes (Puska et al., 2018; Zunke et al., 2018). The resulting neuronal degeneration
process can further lead to the secretion of α-Syn. Glial cells are able to degrade
neurotoxic protein aggregates by their uptake and lysosomal clearance, thus contributing
to neuroprotection. Secreted α-Syn protein aggregates can influence glial cells and
their lysosomal function in certain ways: (1) The uptake of α-Syn aggregates in oligodendrocytes
has been shown to decrease lysosomal CTSD activity (Kaji et al., 2018). (2) Astrocytes
can clear endocytosed α-Syn aggregates in lysosomes, however, simultaneously stimulate
the secretion of astrocytic pro-inflammatory factors like TNFα, IL-1, IL-6, or CXCL1.
These factors induce neuroinflammation and further contribute to neurodegeneration
processes (Kaji et al., 2020). (3) Microglia are critical participants in the immune
response of the CNS by eliminating pathogens and protein aggregates. Microglial α-Syn
uptake can also trigger neuroinflammation in neurodegenerative processes by the transmission
of pro-inflammatory molecules, like IL-1β (Kam et al., 2020; Kreher et al., 2021).
Overall, the lysosomal function within glial cells could contribute towards protecting
neurons from neurotoxic protein aggregates by degrading processes. Nevertheless, glial
clearance capacity of protein aggregates is limited, and an overload of α-Syn accumulation
can promote neuroinflammation. Moreover, it is unclear if and to what extend α-Syn
aggregates within glial cells interfere with general lysosomal function and other
intracellular pathways. Source of images (neurons and glial cells): https://smart.servier.com/.
Role of lysosomes within glial cells: The interaction between neurons and glial cells
is essential for balanced brain homeostasis. Glial cells can be differentiated into
three subtypes: astrocytes, microglia, and oligodendrocytes. Astrocytes, as the most
abundant glial cell type in the CNS, have diverse supporting functions on neurons.
These include nutrition supply, modulation of the blood-brain barrier, immune signaling,
and neurotransmitter recycling. Microglia are CNS-resident macrophages, important
for the immune defense within the brain, whereas oligodendrocytes are specialized
in the synthesis of myelin for axonal insulation, which is crucial for proper neuronal
function (Kreher et al., 2021). Many processes which are vital for glial cell function
involve lysosomal or autophagic pathways and are summarized below for each glial cell
type.
Lysosomes within astrocytes play an important role in membrane recycling, cell signaling,
and clearance of protein compounds. Astrocytes can release proteolytic enzymes and
signal molecules via lysosomal exocytosis. One important signal molecule is the gliotransmitter
ATP, which contributes to the crosstalk between astrocytes and other cells of the
CNS, facilitating neuronal activity as well as synaptic plasticity. The interaction
between neurons and astrocytes becomes further evident in endocytic events: extracellular
protein aggregates (e.g., α-Syn), myelin debris or toxic lipid droplets secreted from
neurons can be taken up by astrocytes and processed within their lysosome. This indicates
a potential molecular mechanism of the CNS to deal with harmful neuronal products
(Kam et al., 2020; Kreher et al., 2021). In microglia, lysosomes are crucial for modulating
synaptic plasticity and immune responses. The exocytosis of the brain-derived neurotrophic
factor plays an important role regarding the development of dendritic spines of neurons.
Furthermore, exocytosis of the acidic hydrolase cathepsin S, as expressed in antigen-presenting
cells like microglia, could also contribute to spine formation by degradation processes
of the extracellular matrix. Endo- and phagocytosis events of microglia are responsible
for the clearance of myelin debris. Additionally, microglial phagocytosis is critical
for the degradation of extracellular aggregates and pathogens, which underlines their
essential role in immune response (Kreher et al., 2021).
Lysosomes within oligodendrocytes play a crucial role in the recycling process of
certain myelin proteins. Myelin serves as an insulating layer by enveloping the axons
of neurons. It consists of different proteins such as myelin basic protein, proteolipid
protein or myelin-associated glycoprotein. Lysosomal exocytosis in oligodendrocytes
has been shown to modulate myelin plasticity by secreting myelin proteins. The protein
lethal giant larvae 1 (Lgl1) is known to mediate vesicular acidification as well as
lysosomal maturation, since knockout (KO) oligodendrocyte precursor cells (OPCs) indicate
abnormal alterations of lysosomal shape. In cell culture and transgenic mouse models,
the deficiency of proteins involved in vesicle transport such as VAMP3/VAMP7, Rab27
or CTSD, can disturb the exocytotic process and thus, lead to an impairment of the
myelination process, often linked to LSDs (Kreher et al., 2021).
Lysosomal pathways in glia associated with synucleinopathies: Recent studies document
the important role of glial cells in the disease progression of PD and MSA. Importantly,
a cell-to-cell transmission of α-Syn released from degenerated neurons to neighboring
glial cells has been proposed. It has been shown that primary glial cells of mouse
models overexpressing wild-type α-Syn, are able to process aggregated α-Syn, contributing
to α-Syn homeostasis and preventing neurodegeneration (Choi et al., 2020).
Although astrocytes express low levels of α-Syn, a deficiency of α-Syn leads to impaired
uptake and trafficking of fatty acids in these glial cells (Kam et al., 2020). The
uptake of neuronal α-Syn in astrocytes occurs via phagocytosis, which has been shown
in a primary astroglial culture. There are several suggestions for possible transfer
mechanisms of neuronal α-Syn between neurons and astrocytes. During oxidative stress,
astrocytes are able to form tunneling nanotubes, which can serve as connection to
other non-stressed cells. Concurrently, the formation of these nanotubes can also
enhance α-Syn spreading. Additionally, the transmission of α-Syn via exosomes comprises
another intracellular mechanism (Mavroeidi and Xilouri, 2021).
The uptake of secreted, neuronal α-Syn species like oligomers or fibrils was demonstrated
in induced pluripotent stem cells-derived astrocytes from a healthy control, suggesting
a protective function towards pathologic α-Syn clearance (
Figure 1
) (Tsunemi et al., 2020). Moreover, investigations from co-cultures of primary astrocytes
with human neuroblastoma cells show, that exogenous α-Syn drives the formation of
inclusion bodies in astrocytes (Kam et al., 2020). The astrocytic uptake of neuron-derived
α-Syn aggregates can also promote the production of astroglial proinflammatory cytokines
(IL-1, IL-6, TNF-α) and chemokines (CXCL1) (
Figure 1
), leading to neuroinflammation contributing to neurodegeneration in PD and MSA (Kaji
et al., 2020; Kam et al., 2020). Therefore, α-Syn can be considered as an exogenous
stimulator of astrocytes (Kam et al., 2020). Furthermore, induced pluripotent stem
cell-derived astroglia carrying GBA1 mutations, showed disturbed lysosomal enzyme
activity and consequently aggregation of α-Syn (Kam et al., 2020). In line with this
data, also GBA1 KO mice demonstrated astroglial activation and abnormal α-Syn accumulation.
Another mouse model deficient for GBA1 within neural and glial progenitor cells exhibited
increased expression of lysosomal cathepsins within astrocytes as well as neurons.
These cathepsins were further distributed to degenerating neurons of affected brain
areas, potentially driving disease pathology (Kam et al., 2020). It is still a matter
of debate if astrocytes drive disease progression or have a neuroprotective effect
within α-synucleinopathies. On the one hand, α-Syn accumulation in astrocytes mediates
inflammatory events due to the secretion of cytokines or chemokines. On the other
hand, aggregates as found within glial cells could be an indication of a neuroprotective
degradation mechanism protecting neuronal cells from toxic protein accumulations (Kaji
et al., 2020).
Interestingly, investigations of brain samples from MSA patients showed, that α-Syn
aggregates, originated from oligodendroglia residues, where engulfed by astrocytes
via phagocytosis and could be detected within astrocytic lysosomes (Puska et al.,
2018). In fact, astrocytes show a higher endocytosis activity and lysosomal proteolysis
compared to neurons, indicating a better capacity to degrade certain substrates, including
α-Syn aggregates (Tsunemi et al., 2020). Therefore, astrocytic degradation processes
regarding α-Syn clearance would comprise an essential therapeutic strategy, especially
in α-synucleinopathies.
So far, there is no clear evidence for endogenous α-Syn expression in microglia. Nevertheless,
a recent study verified the clearance of neuronal α-Syn by the autophagy-lysosomal
pathway (
Figure 1
), which indicates the important role of microglial neuroprotection (Choi et al.,
2020). Moreover, microglia were able to take up α-Syn from exosomes released from
oligodendrocytes (Kaji et al., 2020). However, pathologic α-Syn species, like oligomers
or fibrils, can activate several microglial receptors and function as damage-associated
molecular patterns (DAMPs). For instance, α-Syn can reduce microglial phagocytosis
by binding on the surface receptor FcγRIIB and consequently, disturbs the clearance
mechanisms of aggregated species or cell debris. Furthermore, fibrillar α-Syn can
induce a series of pro-inflammatory events by activating the nuclear factor-kappa
B (NF-κB) pathway, which is crucial for microglial inflammatory response. The subsequent
release of microglial cytokines (e.g., IL-1β) contributes to neuroinflammation in
PD (Kam et al., 2020). It is suggested that neuronal α-Syn can be phagocytosed by
microglia via the lymphocyte-activation gene 3 (LAG3), and could further transmit
α-Syn aggregates due to disturbed lysosomal clearance and exocytosis, which in turn
induces pro-inflammatory microglial response (
Figure 1
; Kreher et al., 2021). Plasma levels of PD patients, carrying mutations in the coding
gene for the lysosomal enzyme GBA1, were shown to have increased cytokine and inflammatory
markers and promote microglia-mediated neuronal dysfunction (Kam et al., 2020). Surprisingly,
the microglial β-glucocerebrosidase (GBA1) function has not been investigated on a
mechanistic level to date.
Observations in MSA brains show a higher microglia cell density with increasing degeneration
of neurons, possibly indicating a higher proliferation of microglia during neuroinflammation.
With a high migration capacity, microglia possibly accelerate the formation and distribution
of α-Syn aggregation and could promote α-Syn transmission by its uptake and release
(Kaji et al., 2020).
Oligodendrocytes have been described to express low levels of endogenous α-Syn compared
to neurons (Kaji et al., 2020). Accumulations of fibrillar α-Syn in oligodendrocytes
are prevalent constituents of glial cytoplasmic inclusions in brain regions of MSA
patients. Interestingly, an in vitro experiment in primary oligodendrocyte lineage
cell cultures revealed, that external α-Syn fibrils did not affect the expression
level of lysosomal enzymes, e.g., CTSD in OPCs and mature oligodendrocytes. However,
α-Syn aggregates were able to diminish CTSD enzyme activity, especially in OPCs (
Figure 1
; Kaji et al., 2018). Importantly, CTSD deficiency in a transgenic mouse model has
been shown to delay myelin maturation and oligodendrocyte development, underlining
the importance of CTSD in oligodendrocyte function (Guo et al., 2018). Interestingly,
in the terminal pathological phase of MSA, oligodendrocytes rarely harbor lysosomal
α-Syn in comparison to PD, where lysosomal α-Syn can be found in neurons (Puska et
al., 2018). Surprisingly, the function and homeostasis of oligodendrocytes during
disease progression in PD still remains elusive. In the future, the impact of OPCs
and oligodendrocytes in α-synucleinopathy has to be investigated in more detail, allowing
a better understanding of oligodendroglial alteration in α-Syn-related pathology.
Overall, α-Syn formations can trigger specific responses in the individual glial cells,
which might contribute to either neuroprotection by lysosomal α-Syn degradation or
further drive disease progression in neurodegeneration. This clearly underlines the
importance of an unimpaired glial functionality with a special focus on lysosomal
pathways.
Perspective: As depicted in this work, there is still a large lack of knowledge about
the role of lysosomal pathways within glial cells under pathological, but also physiological
conditions. In order to better understand the whole picture of disease pathways resulting
in α-synucleinopathies, glial cell biology has to be studied in more detail. For instance,
it is unknown if intracellular disease mechanisms, as found in neurons, recapitulate
within glial cells. It would be interesting to know if α-Syn aggregates that have
been shown to impact lysosomal function within neurons, for example by interfering
with intracellular protein transport, exert the same effects within glia. Moreover,
further studies will need to address the consequences of PD-associated genetic variants
within lysosome-associated proteins in glial cells and how this compares to neurons.
These future investigations are needed for a more detailed knowledge about the exact
pathways of α-Syn uptake and clearance in glial cells to enable new therapeutic strategies
in neurodegenerative diseases.
We thank Yanni Schneider, Alice Drobny and Susy Prieto Huarcaya for critically proofreading
the manuscript.
This work was supported by the Interdisciplinary Center for Clinical Research (IZKF)
at the University Hospital of the University of Erlangen-Nuremberg (Jochen-Kalden
funding programme N8).
Related collections
Most cited references13

- Record: found
- Abstract: found
- Article: found
Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration
Insup Choi, Yuanxi Zhang, Steven Seegobin … (2020)
- Record: found
- Abstract: found
- Article: not found
Determinants of dopaminergic neuron loss in Parkinson's disease
Dalton Surmeier (2018)
- Record: found
- Abstract: not found
- Article: not found
Microglia and astrocyte dysfunction in parkinson's disease
Tae-In Kam, Jared T Hinkle, Ted Dawson … (2020)