- Record: found
- Abstract: found
- Article: found
Expression profiling of white sponge nevus by RNA sequencing revealed pathological pathways
Read this article at
Abstract
Background
White sponge nevus (WSN) is a rare periodontal hereditary disease. To date, almost all WSN studies have focused on case reports or mutation reports. Thus, the mechanism behind WSN is still unclear. We investigated the pathogenesis of WSN using expression profiling.
Methods
Sequence analysis of samples from a WSN Chinese family revealed a mutation (332 T > C) in the KRT13 gene that resulted in the amino acid change Leu111Pro. The pathological pathway behind the WSN expression profile was investigated by RNA sequencing (RNA-seq).
Results
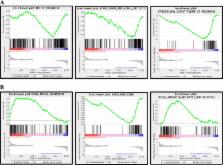
Construction of a heatmap revealed 24 activated genes and 57 reduced genes in the WSN patients. The ribosome structure was damaged in the WSN patients. Moreover, the translation rate was limited in the WSN patients, whereas ubiquitin-mediated proteolysis was enhanced.
Related collections
Most cited references30
- Record: found
- Abstract: found
- Article: not found
The transcriptional landscape of the yeast genome defined by RNA sequencing.
- Record: found
- Abstract: found
- Article: not found
The impact of next-generation sequencing technology on genetics.
- Record: found
- Abstract: found
- Article: not found