- Record: found
- Abstract: found
- Article: found
MTBseq: a comprehensive pipeline for whole genome sequence analysis of Mycobacterium tuberculosis complex isolates
Read this article at
Abstract
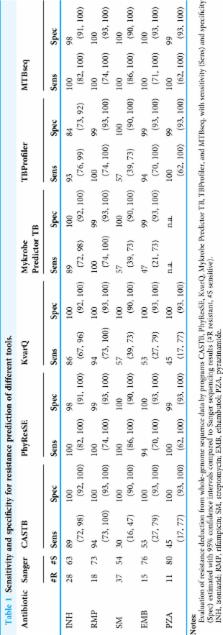
Analyzing whole-genome sequencing data of Mycobacterium tuberculosis complex (MTBC) isolates in a standardized workflow enables both comprehensive antibiotic resistance profiling and outbreak surveillance with highest resolution up to the identification of recent transmission chains. Here, we present MTBseq, a bioinformatics pipeline for next-generation genome sequence data analysis of MTBC isolates. Employing a reference mapping based workflow, MTBseq reports detected variant positions annotated with known association to antibiotic resistance and performs a lineage classification based on phylogenetic single nucleotide polymorphisms (SNPs). When comparing multiple datasets, MTBseq provides a joint list of variants and a FASTA alignment of SNP positions for use in phylogenomic analysis, and identifies groups of related isolates. The pipeline is customizable, expandable and can be used on a desktop computer or laptop without any internet connection, ensuring mobile usage and data security. MTBseq and accompanying documentation is available from https://github.com/ngs-fzb/MTBseq_source.
Related collections
Most cited references14
- Record: found
- Abstract: found
- Article: not found
The Bioperl toolkit: Perl modules for the life sciences.
- Record: found
- Abstract: found
- Article: not found
Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage.

- Record: found
- Abstract: found
- Article: found