- Record: found
- Abstract: found
- Article: found
Molecular Landscapes and Models of Acute Erythroleukemia
Read this article at
Abstract
Supplemental Digital Content is available in the text.
Abstract
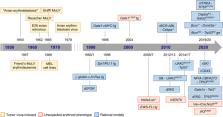
Malignancies of the erythroid lineage are rare but aggressive diseases. Notably, the first insights into their biology emerged over half a century ago from avian and murine tumor viruses-induced erythroleukemia models providing the rationale for several transgenic mouse models that unraveled the transforming potential of signaling effectors and transcription factors in the erythroid lineage. More recently, genetic roadmaps have fueled efforts to establish models that are based on the epigenomic lesions observed in patients with erythroid malignancies. These models, together with often unexpected erythroid phenotypes in genetically modified mice, provided further insights into the molecular mechanisms of disease initiation and maintenance. Here, we review how the increasing knowledge of human erythroleukemia genetics combined with those from various mouse models indicate that the pathogenesis of the disease is based on the interplay between signaling mutations, impaired TP53 function, and altered chromatin organization. These alterations lead to aberrant activity of erythroid transcriptional master regulators like GATA1, indicating that erythroleukemia will most likely require combinatorial targeting for efficient therapeutic interventions.
Related collections
Most cited references171
- Record: found
- Abstract: found
- Article: not found
The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia.
- Record: found
- Abstract: found
- Article: not found
Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia.
- Record: found
- Abstract: found
- Article: not found