- Record: found
- Abstract: found
- Article: found
Genetic basis and adaptation trajectory of soybean from its temperate origin to tropics
Read this article at
Abstract
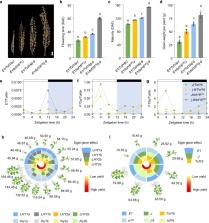
Soybean ( Glycine max) serves as a major source of protein and edible oils worldwide. The genetic and genomic bases of the adaptation of soybean to tropical regions remain largely unclear. Here, we identify the novel locus Time of Flowering 16 ( Tof16), which confers delay flowering and improve yield at low latitudes and determines that it harbors the soybean homolog of LATE ELONGATED HYPOCOTYL ( LHY). Tof16 and the previously identified J locus genetically additively but independently control yield under short-day conditions. More than 80% accessions in low latitude harbor the mutations of tof16 and j, which suggests that loss of functions of Tof16 and J are the major genetic basis of soybean adaptation into tropics. We suggest that maturity and yield traits can be quantitatively improved by modulating the genetic complexity of various alleles of the LHY homologs, J and E1. Our findings uncover the adaptation trajectory of soybean from its temperate origin to the tropics.
Abstract
How soybean, a temperate origin crop, adapted to a tropical environment remains unclear. Here, the authors report Tof16, an ortholog of LHY, and the previously identified J locus, control soybean yield under short-day condition and loss of function of these two genes contributes to the adaptation to tropics.
Related collections
Most cited references38

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools

- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform
- Record: found
- Abstract: found
- Article: not found