- Record: found
- Abstract: found
- Article: not found
Plasmodium falciparum Histones Induce Endothelial Proinflammatory Response and Barrier Dysfunction
Read this article at

Abstract
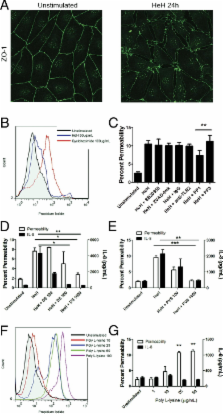
Plasmodium falciparum is a protozoan parasite of human erythrocytes that causes the most severe form of malaria. Severe P. falciparum infection is associated with endothelial activation and permeability, which are important determinants of the outcome of the infection. How endothelial cells become activated is not fully understood, particularly with regard to the effects of parasite subcomponents. We demonstrated that P. falciparum histones extracted from merozoites (HeH) directly stimulated the production of IL-8 and other inflammatory mediators by primary human dermal microvascular endothelial cells through a signaling pathway that involves Src family kinases and p38 MAPK. The stimulatory effect of HeH and recombinant P. falciparum H3 (PfH3) was abrogated by histone-specific antibodies. The release of nuclear contents on rupture of infected erythrocytes was captured by live cell imaging and confirmed by detecting nucleosomes in the supernatants of parasite cultures. HeH and recombinant parasite histones also induced endothelial permeability through a charge-dependent mechanism that resulted in disruption of junctional protein expression and cell death. Recombinant human activated protein C cleaved HeH and PfH3 and abrogated their proinflammatory effects. Circulating nucleosomes of both human and parasite origin were detected in the plasma of patients with falciparum malaria and correlated positively with disease severity. These results support a pathogenic role for both host- and pathogen-derived histones in P. falciparum-caused malaria.
Related collections
Most cited references51
- Record: found
- Abstract: found
- Article: not found
Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9.
- Record: found
- Abstract: found
- Article: not found
Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin
- Record: found
- Abstract: found
- Article: not found