- Record: found
- Abstract: found
- Article: found
HaplotypeTools: a toolkit for accurately identifying recombination and recombinant genotypes
Read this article at
Abstract
Background
Identifying haplotypes is central to sequence analysis in diploid or polyploid genomes. Despite this, there remains a lack of research and tools designed for physical phasing and its downstream analysis.
Results
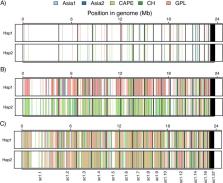
HaplotypeTools is a new toolset to phase variant sites using VCF and BAM files and to analyse phased VCFs. Phasing is achieved via the identification of reads overlapping ≥ 2 heterozygous positions and then extended by additional reads, a process that can be parallelized across a computer cluster. HaplotypeTools includes various utility scripts for downstream analysis including crossover detection and phylogenetic placement of haplotypes to other lineages or species. HaplotypeTools was assessed for accuracy against WhatsHap using simulated short and long reads, demonstrating higher accuracy, albeit with reduced haplotype length. HaplotypeTools was also tested on real Illumina data to determine the ancestry of hybrid fungal isolate Batrachochytrium dendrobatidis ( Bd) SA-EC3, finding 80% of haplotypes across the genome phylogenetically cluster with parental lineages BdGPL (39%) and BdCAPE (41%), indicating those are the parental lineages. Finally, ~ 99% of phasing was conserved between overlapping phase groups between SA-EC3 and either parental lineage, indicating mitotic gene conversion/parasexuality as the mechanism of recombination for this hybrid isolate. HaplotypeTools is open source and freely available from https://github.com/rhysf/HaplotypeTools under the MIT License.
Related collections
Most cited references29

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools
- Record: found
- Abstract: found
- Article: not found
The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data.

- Record: found
- Abstract: found
- Article: found