- Record: found
- Abstract: found
- Article: found
The huntingtin N17 domain is a multifunctional CRM1 and Ran-dependent nuclear and cilial export signal
Read this article at
Abstract
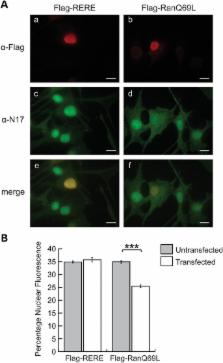
The first 17 amino acids of Huntington’s disease (HD) protein, huntingtin, comprise an amphipathic alpha-helical domain that can target huntingtin to the endoplasmic reticulum (ER). N17 is phosphorylated at two serines, shown to be important for disease development in genetic mouse models, and shown to be modified by agents that reverse the disease phenotype in an HD mouse model. Here, we show that the hydrophobic face of N17 comprises a consensus CRM1/exportin-dependent nuclear export signal, and that this nuclear export activity can be affected by serine phospho-mimetic mutants. We define the precise residues that comprise this nuclear export sequence (NES) as well as the interaction of the NES, but not phospho-mimetic mutants, with the CRM1 nuclear export factor. We show that the nuclear localization of huntingtin depends upon the RanGTP/GDP gradient, and that N17 phosphorylation can also distinguish localization of endogenous huntingtin between the basal body and stalk of the primary cilium. We present a mechanism and multifunctional role for N17 in which phosphorylation of N17 not only releases huntingtin from the ER to allow nuclear entry, but also prevents nuclear export during a transient stress response event to increase the levels of nuclear huntingtin and to regulate huntingtin access to the primary cilium. Thus, N17 is a master localization signal of huntingtin that can mediate huntingtin localization between the cytoplasm, nucleus and primary cilium. This localization can be regulated by signaling, and is misregulated in HD.
Related collections
Most cited references49
- Record: found
- Abstract: found
- Article: not found
Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice.
- Record: found
- Abstract: found
- Article: not found
Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells.
- Record: found
- Abstract: found
- Article: not found