- Record: found
- Abstract: found
- Article: found
Zelda is differentially required for chromatin accessibility, transcription factor binding, and gene expression in the early Drosophila embryo
Read this article at
Abstract
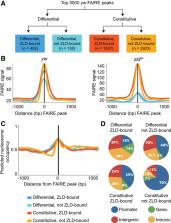
The transition from a specified germ cell to a population of pluripotent cells occurs rapidly following fertilization. During this developmental transition, the zygotic genome is largely transcriptionally quiescent and undergoes significant chromatin remodeling. In Drosophila, the DNA-binding protein Zelda (also known as Vielfaltig) is required for this transition and for transcriptional activation of the zygotic genome. Open chromatin is associated with Zelda-bound loci, as well as more generally with regions of active transcription. Nonetheless, the extent to which Zelda influences chromatin accessibility across the genome is largely unknown. Here we used formaldehyde-assisted isolation of regulatory elements to determine the role of Zelda in regulating regions of open chromatin in the early embryo. We demonstrate that Zelda is essential for hundreds of regions of open chromatin. This Zelda-mediated chromatin accessibility facilitates transcription-factor recruitment and early gene expression. Thus, Zelda possesses some key characteristics of a pioneer factor. Unexpectedly, chromatin at a large subset of Zelda-bound regions remains open even in the absence of Zelda. The GAGA factor-binding motif and embryonic GAGA factor binding are specifically enriched in these regions. We propose that both Zelda and GAGA factor function to specify sites of open chromatin and together facilitate the remodeling of the early embryonic genome.
Related collections
Most cited references72

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools
- Record: found
- Abstract: found
- Article: not found
Fast gapped-read alignment with Bowtie 2.

- Record: found
- Abstract: found
- Article: found