- Record: found
- Abstract: found
- Article: found
Duplication and Specialization of NUDX1 in Rosaceae Led to Geraniol Production in Rose Petals
Read this article at
Abstract
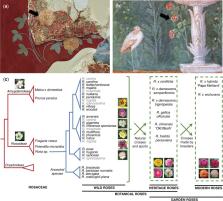
Nudix hydrolases are conserved enzymes ubiquitously present in all kingdoms of life. Recent research revealed that several Nudix hydrolases are involved in terpenoid metabolism in plants. In modern roses, RhNUDX1 is responsible for formation of geraniol, a major compound of rose scent. Nevertheless, this compound is produced by monoterpene synthases in many geraniol-producing plants. As a consequence, this raised the question about the origin of RhNUDX1 function and the NUDX1 gene evolution in Rosaceae, in wild roses or/and during the domestication process. Here, we showed that three distinct clades of NUDX1 emerged in the Rosoidae subfamily (Nudx1-1 to Nudx1-3 clades), and two subclades evolved in the Rosa genus (Nudx1-1a and Nudx1-1b subclades). We also showed that the Nudx1-1b subclade was more ancient than the Nudx1-1a subclade, and that the NUDX1-1a gene emerged by a trans-duplication of the more ancient NUDX1-1b gene. After the transposition, NUDX1-1a was cis-duplicated, leading to a gene dosage effect on the production of geraniol in different species. Furthermore, the NUDX1-1a appearance was accompanied by the evolution of its promoter, most likely from a Copia retrotransposon origin, leading to its petal-specific expression. Thus, our data strongly suggest that the unique function of NUDX1-1a in geraniol formation was evolved naturally in the genus Rosa before domestication.
Related collections
Most cited references72

- Record: found
- Abstract: found
- Article: found
BLAST+: architecture and applications
- Record: found
- Abstract: found
- Article: not found
A new mathematical model for relative quantification in real-time RT-PCR.
- Record: found
- Abstract: found
- Article: not found