- Record: found
- Abstract: found
- Article: found
Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis
Read this article at
Abstract
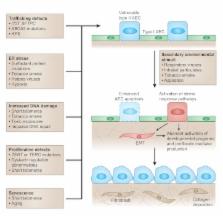
Idiopathic pulmonary fibrosis (IPF) is a progressive and often fatal lung disease for which there is no known treatment. Although the traditional paradigm of IPF pathogenesis emphasized chronic inflammation as the primary driver of fibrotic remodeling, more recent insights have challenged this view. Linkage analysis and candidate gene approaches have identified four genes that cause the inherited form of IPF, familial interstitial pneumonia (FIP). These four genes encode two surfactant proteins, surfactant protein C (encoded by SFTPC) and surfactant protein A2 ( SFTPA2), and two components of the telomerase complex, telomerase reverse transcriptase ( TERT) and the RNA component of telomerase ( TERC). In this review, we discuss how investigating these mutations, as well as genetic variants identified in other inherited disorders associated with pulmonary fibrosis, are providing new insights into the pathogenesis of common idiopathic interstitial lung diseases, particularly IPF. Studies in this area have highlighted key roles for epithelial cell injury and dysfunction in the development of lung fibrosis. In addition, genetic approaches have uncovered the importance of several processes – including endoplasmic reticulum stress and the unfolded protein response, DNA-damage and -repair pathways, and cellular senescence – that might provide new therapeutic targets in fibrotic lung diseases.
Related collections
Most cited references70
- Record: found
- Abstract: found
- Article: not found
Telomere dysfunction induces metabolic and mitochondrial compromise.
- Record: found
- Abstract: found
- Article: not found
A common MUC5B promoter polymorphism and pulmonary fibrosis.
- Record: found
- Abstract: found
- Article: not found