- Record: found
- Abstract: found
- Article: found
Profiling the Structural Determinants of Aryl Benzamide Derivatives as Negative Allosteric Modulators of mGluR5 by In Silico Study
Read this article at
Abstract
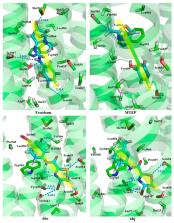
Glutamate plays a crucial role in the treatment of depression by interacting with the metabotropic glutamate receptor subtype 5 (mGluR5), whose negative allosteric modulators (NAMs) are thus promising antidepressants. At present, to explore the structural features of 106 newly synthesized aryl benzamide series molecules as mGluR5 NAMs, a set of ligand-based three-dimensional quantitative structure-activity relationship (3D-QSAR) analyses were firstly carried out applying comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) methods. In addition, receptor-based analysis, namely molecular docking and molecular dynamics (MD) simulations, were performed to further elucidate the binding modes of mGluR5 NAMs. As a result, the optimal CoMSIA model obtained shows that cross-validated correlation coefficient Q 2 = 0.70, non-cross-validated correlation coefficient R 2 ncv = 0.89, predicted correlation coefficient R 2 pre = 0.87. Moreover, we found that aryl benzamide series molecules bind as mGluR5 NAMs at Site 1, which consists of amino acids Pro655, Tyr659, Ile625, Ile651, Ile944, Ser658, Ser654, Ser969, Ser965, Ala970, Ala973, Trp945, Phe948, Pro903, Asn907, Val966, Leu904, and Met962. This site is the same as that of other types of NAMs; mGluR5 NAMs are stabilized in the “linear” and “arc” configurations mainly through the H-bonds interactions, π–π stacking interaction with Trp945, and hydrophobic contacts. We hope that the models and information obtained will help understand the interaction mechanism of NAMs and design and optimize NAMs as new types of antidepressants.
Related collections
Most cited references30
- Record: found
- Abstract: found
- Article: not found
CHARMM-GUI: a web-based graphical user interface for CHARMM.
- Record: found
- Abstract: not found
- Article: not found
Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins.
- Record: found
- Abstract: not found
- Article: not found