- Record: found
- Abstract: found
- Article: found
Phenotypic screen for oxygen consumption rate identifies an anti-cancer naphthoquinone that induces mitochondrial oxidative stress

Read this article at
Abstract
A hallmark of cancer cells is their ability to reprogram nutrient metabolism. Thus, disruption to this phenotype is a potential avenue for anti-cancer therapy. Herein we used a phenotypic chemical library screening approach to identify molecules that disrupted nutrient metabolism (by increasing cellular oxygen consumption rate) and were toxic to cancer cells. From this screen we discovered a 1,4-Naphthoquinone (referred to as BH10) that is toxic to a broad range of cancer cell types. BH10 has improved cancer-selective toxicity compared to doxorubicin, 17-AAG, vitamin K3, and other known anti-cancer quinones. BH10 increases glucose oxidation via both mitochondrial and pentose phosphate pathways, decreases glycolysis, lowers GSH:GSSG and NAPDH/NAPD + ratios exclusively in cancer cells, and induces necrosis. BH10 targets mitochondrial redox defence as evidenced by increased mitochondrial peroxiredoxin 3 oxidation and decreased mitochondrial aconitase activity, without changes in markers of cytosolic or nuclear damage. Over-expression of mitochondria-targeted catalase protects cells from BH10-mediated toxicity, while the thioredoxin reductase inhibitor auranofin synergistically enhances BH10-induced peroxiredoxin 3 oxidation and cytotoxicity. Overall, BH10 represents a 1,4-Naphthoquinone with an improved cancer-selective cytotoxicity profile via its mitochondrial specificity.
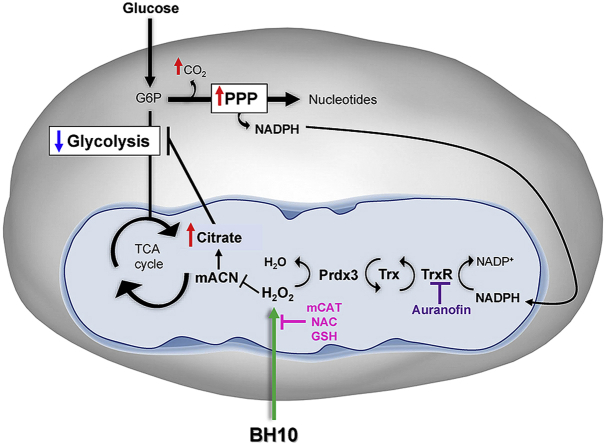
Graphical abstract
Schematic overview of BH10-induced effects on cancer cells. Treatment of cancer cells with BH10 induced mitochondrial oxidative stress. Activity of the redox-sensitive enzyme mitochondrial aconitase (mACN) was impaired leading to citrate accumulation and a decrease in glycolysis. BH10 increased pentose phosphate pathway activity to increase NADPH production for antioxidant defence. Mitochondrial oxidative stress is evidenced by oxidation of the peroxiredoxin/thioredoxin reductase system, including peroxiredoxin 3 dimerization (oxidation), and depletion of NADPH. BH10's cytotoxicity and effect on PRDX3 dimerization were synergistically enhanced with the thioredoxin reductase inhibitor auranofin, while over-expression of mitochondrial-targeted catalase (mCAT) or treatment with antioxidants N-acetylcysteine (NAC) and GSH protected cells from BH10-induced death.
Highlights
Related collections
Most cited references22

- Record: found
- Abstract: found
- Article: found
Glutathione metabolism in cancer progression and treatment resistance
- Record: found
- Abstract: found
- Article: not found
Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer
- Record: found
- Abstract: found
- Article: not found