- Record: found
- Abstract: found
- Article: found
R-loops and nicks initiate DNA breakage and genome instability in non-growing Escherichia coli
Read this article at
Abstract
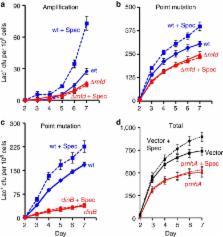
Double-stranded DNA ends, often from replication, drive genomic instability, yet their origin in non-replicating cells is unknown. Here we show that transcriptional RNA/DNA hybrids (R-loops) generate DNA ends that underlie stress-induced mutation and amplification. Depleting RNA/DNA hybrids with overproduced RNase HI reduces both genomic changes, indicating RNA/DNA hybrids as intermediates in both. An Mfd requirement and inhibition by translation implicate transcriptional R-loops. R-loops promote instability by generating DNA ends, shown by their dispensability when ends are provided by I- SceI endonuclease. Both R-loops and single-stranded endonuclease TraI are required for end formation, visualized as foci of a fluorescent end-binding protein. The data suggest that R-loops prime replication forks that collapse at single-stranded nicks, producing ends that instigate genomic instability. The results illuminate how DNA ends form in non-replicating cells, identify R-loops as the earliest known mutation/amplification intermediate, and suggest that genomic instability during stress could be targeted to transcribed regions, accelerating adaptation.
Abstract
 DNA double-strand breaks commonly occur in all replicating cells. Wimberly and colleagues
show that in non-replicating cells, aborted transcription/translation forms RNA/DNA
hybrid R-loops that prime origin-independent replication, leading to DNA breakage,
point mutations and chromosomal rearrangements.
DNA double-strand breaks commonly occur in all replicating cells. Wimberly and colleagues
show that in non-replicating cells, aborted transcription/translation forms RNA/DNA
hybrid R-loops that prime origin-independent replication, leading to DNA breakage,
point mutations and chromosomal rearrangements.
Related collections
Most cited references46
- Record: found
- Abstract: found
- Article: not found
Mechanisms of change in gene copy number.
- Record: found
- Abstract: found
- Article: not found
Single-strand break repair and genetic disease.
- Record: found
- Abstract: found
- Article: not found