- Record: found
- Abstract: found
- Article: not found
Testing of the GROMOS Force-Field Parameter Set 54A8: Structural Properties of Electrolyte Solutions, Lipid Bilayers, and Proteins
Read this article at

Abstract
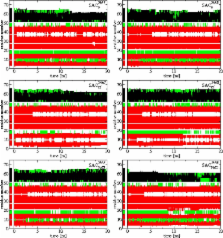
The GROMOS 54A8 force field [Reif et al. J. Chem. Theory Comput. 2012, 8, 3705–3723] is the first of its kind to contain nonbonded parameters for charged amino acid side chains that are derived in a rigorously thermodynamic fashion, namely a calibration against single-ion hydration free energies. Considering charged moieties in solution, the most decisive signature of the GROMOS 54A8 force field in comparison to its predecessor 54A7 can probably be found in the thermodynamic equilibrium between salt-bridged ion pair formation and hydration. Possible shifts in this equilibrium might crucially affect the properties of electrolyte solutions or/and the stability of (bio)molecules. It is therefore important to investigate the consequences of the altered description of charged oligoatomic species in the GROMOS 54A8 force field. The present study focuses on examining the ability of the GROMOS 54A8 force field to accurately model the structural properties of electrolyte solutions, lipid bilayers, and proteins. It is found that ( i) aqueous electrolytes involving oligoatomic species (sodium acetate, methylammonium chloride, guanidinium chloride) reproduce experimental salt activity derivatives for concentrations up to 1.0 m (1.0-molal) very well, and good agreement between simulated and experimental data is also reached for sodium acetate and methylammonium chloride at 2.0 m concentration, while not even qualitative agreement is found for sodium chloride throughout the whole range of examined concentrations, indicating a failure of the GROMOS 54A7 and 54A8 force-field parameter sets to correctly account for the balance between ion–ion and ion–water binding propensities of sodium and chloride ions; ( ii) the GROMOS 54A8 force field reproduces the liquid crystalline-like phase of a hydrated DPPC bilayer at a pressure of 1 bar and a temperature of 323 K, the area per lipid being in agreement with experimental data, whereas other structural properties (volume per lipid, bilayer thickness) appear underestimated; ( iii) the secondary structure of a range of different proteins simulated with the GROMOS 54A8 force field at pH 7 is maintained and compatible with experimental NMR data, while, as also observed for the GROMOS 54A7 force field, α-helices are slightly overstabilized with respect to 3 10-helices; ( iv) with the GROMOS 54A8 force field, the side chains of arginine, lysine, aspartate, and glutamate residues appear slightly more hydrated and present a slight excess of oppositely-charged solution components in their vicinity, whereas salt-bridge formation properties between charged residues at the protein surface, as assessed by probability distributions of interionic distances, are largely equivalent in the GROMOS 54A7 and 54A8 force-field parameter sets.
Related collections
Most cited references46
- Record: found
- Abstract: not found
- Article: not found
Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features.
- Record: found
- Abstract: found
- Article: found
Why do ultrasoft repulsive particles cluster and crystallize? Analytical results from density functional theory
- Record: found
- Abstract: found
- Article: found