- Record: found
- Abstract: found
- Article: found
Deciphering the potential of a plant growth promoting endophyte Rhizobium sp. WYJ-E13, and functional annotation of the genes involved in the metabolic pathway
Read this article at
Abstract
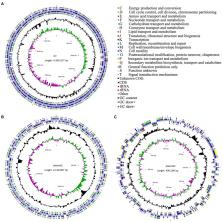
Plant growth-promoting rhizobacteria (PGPR) are well-acknowledged root endophytic bacteria used for plant growth promotion. However, which metabolites produced by PGPR could promote plant growth remains unclear. Additionally, which genes are responsible for plant growth-promoting traits is also not elucidated. Thus, as comprehensive understanding of the mechanism of endophyte in growth promotion is limited, this study aimed to determine the metabolites and genes involved in plant growth-promotion. We isolated an endophytic Rhizobium sp. WYJ-E13 strain from the roots of Curcuma wenyujin Y.H. Chen et C. Ling, a perennial herb and medicinal plant. The tissue culture experiment showed its plant growth-promoting ability. The bacterium colonization in the root was confirmed by scanning electron microscopy and paraffin sectioning. Furthermore, it was noted that the WYJ-E13 strain produced cytokinin, anthranilic acid, and L-phenylalanine by metabolome analysis. Whole-genome analysis of the strain showed that it consists of a circular chromosome of 4,350,227 bp with an overall GC content of 60.34%, of a 2,149,667 bp plasmid1 with 59.86% GC, and of a 406,180 bp plasmid2 with 58.05% GC. Genome annotation identified 4,349 putative protein-coding genes, 51 tRNAs, and 9 rRNAs. The CDSs number allocated to the Kyoto Encyclopedia of Genes and Genomes, Gene Ontology, and Clusters of Orthologous Genes databases were 2027, 3,175 and 3,849, respectively. Comparative genome analysis displayed that Rhizobium sp. WYJ-E13 possesses the collinear region among three species: Rhizobium acidisoli FH23, Rhizobium gallicum R602 and Rhizobium phaseoli R650. We recognized a total set of genes that are possibly related to plant growth promotion, including genes involved in nitrogen metabolism ( nifU, gltA, gltB, gltD, glnA, glnD), hormone production ( trp ABCDEFS), sulfur metabolism ( cysD, cysE, cysK, cysN), phosphate metabolism ( pstA, pstC, phoB, phoH, phoU), and root colonization. Collectively, these findings revealed the roles of WYJ-E13 strain in plant growth-promotion. To the best of our knowledge, this was the first study using whole-genome sequencing for Rhizobium sp. WYJ-E13 associated with C. wenyujin. WYJ-E13 strain has a high potential to be used as Curcuma biofertilizer for sustainable agriculture.
Related collections
Most cited references92
- Record: found
- Abstract: found
- Article: not found
SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing.
- Record: found
- Abstract: found
- Article: not found
Fast and sensitive protein alignment using DIAMOND.

- Record: found
- Abstract: found
- Article: found