
- Record: found
- Abstract: found
- Article: found
Characterizing Microbiomes via Sequencing of Marker Loci: Techniques To Improve Throughput, Account for Cross-Contamination, and Reduce Cost
Read this article at
ABSTRACT
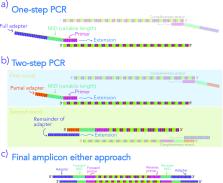
New approaches to characterizing microbiomes via high-throughput sequencing provide impressive gains in efficiency and cost reduction compared to approaches that were standard just a few years ago. However, the speed of method development has been such that staying abreast of the latest technological advances is challenging. Moreover, shifting laboratory protocols to include new methods can be expensive and time consuming. To facilitate adoption of new techniques, we provide a guide and review of recent advances that are relevant for single-locus sequence-based study of microbiomes—from extraction to library preparation—including a primer regarding the use of liquid-handling automation in small-scale academic settings. Additionally, we describe several amendments to published techniques to improve throughput, track contamination, and reduce cost. Notably, we suggest adding synthetic DNA molecules to each sample during nucleic acid extraction, thus providing a method of documenting incidences of cross-contamination. We also describe a dual-indexing scheme for Illumina sequencers that allows multiplexing of many thousands of samples with minimal PhiX input. Collectively, the techniques that we describe demonstrate that laboratory technology need not impose strict limitations on the scale of molecular microbial ecology studies.
IMPORTANCE New methods to characterize microbiomes reduce technology-imposed limitations to study design, but many new approaches have not been widely adopted. Here, we present techniques to increase throughput and reduce contamination alongside a thorough review of current best practices.
Related collections
Most cited references125
- Record: found
- Abstract: found
- Article: not found
Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences
- Record: found
- Abstract: found
- Article: not found
Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB
- Record: found
- Abstract: found
- Article: not found
Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform.
Author and article information
Comments
Comment on this article
See how this article has been cited at scite.ai
scite shows how a scientific paper has been cited by providing the context of the citation, a classification describing whether it supports, mentions, or contrasts the cited claim, and a label indicating in which section the citation was made.