- Record: found
- Abstract: found
- Article: found
Spred2 Deficiency Exacerbates D-Galactosamine/Lipopolysaccharide -induced Acute Liver Injury in Mice via Increased Production of TNFα
Read this article at
Abstract
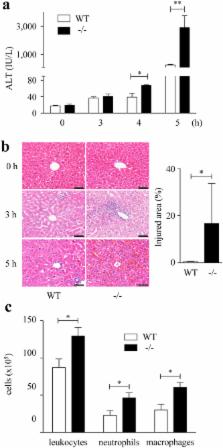
Acute liver injury (ALI) is characterized by hepatocyte damage and inflammation. In the present study, we examined whether the absence of Sprouty-related EVH1-domain-containing protein 2 (Spred2), a negative regulator of the Ras/Raf/ERK/MAPK pathway, influences ALI induced by D-galactosamine (D-GalN) and lipopolysaccharide (LPS). Compared to wild-type mice, Spred2 −/− mice developed exacerbated liver injury represented by enhanced hepatocyte damage and inflammation. Enhanced ERK activation was observed in Spred2 −/−-livers, and the MEK/ERK inhibitor U0126 ameliorated ALI. Hepatic tumour necrosis factor α (TNFα) and interleukin (IL)-1β levels were increased in Spred-2 −/−-livers, and the neutralization of TNFα dramatically ameliorated ALI, which was associated with decreased levels of endogenous TNFα and IL-1β. When mice were challenged with D-GalN and TNFα, much severer ALI was observed in Spred2 −/− mice with significant increases in endogenous TNFα and IL-1β in the livers. Immunohistochemically, Kupffer cells were found to produce TNFα, and isolated Kupffer cells from Spred2 −/− mice produced significantly higher levels of TNFα than those from wild-type mice after LPS stimulation, which was significantly decreased by U0126. These results suggest that Spred2 negatively regulates D-GalN/LPS-induced ALI under the control of TNFα in Kupffer cells. Spred2 may present a therapeutic target for the treatment of ALI.
Related collections
Most cited references34
- Record: found
- Abstract: found
- Article: not found
Mitogen-activated protein kinases in innate immunity.
- Record: found
- Abstract: found
- Article: not found
Identification of a novel inhibitor of mitogen-activated protein kinase kinase.
- Record: found
- Abstract: not found
- Article: not found