- Record: found
- Abstract: found
- Article: found
The Role of the Antioxidant Response in Mitochondrial Dysfunction in Degenerative Diseases: Cross-Talk between Antioxidant Defense, Autophagy, and Apoptosis
Read this article at
Abstract
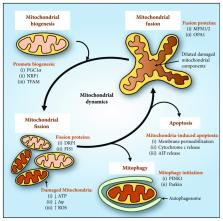
The mitochondrion is an essential organelle important for the generation of ATP for cellular function. This is especially critical for cells with high energy demands, such as neurons for signal transmission and cardiomyocytes for the continuous mechanical work of the heart. However, deleterious reactive oxygen species are generated as a result of mitochondrial electron transport, requiring a rigorous activation of antioxidative defense in order to maintain homeostatic mitochondrial function. Indeed, recent studies have demonstrated that the dysregulation of antioxidant response leads to mitochondrial dysfunction in human degenerative diseases affecting the nervous system and the heart. In this review, we outline and discuss the mitochondrial and oxidative stress factors causing degenerative diseases, such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, Huntington's disease, and Friedreich's ataxia. In particular, the pathological involvement of mitochondrial dysfunction in relation to oxidative stress, energy metabolism, mitochondrial dynamics, and cell death will be explored. Understanding the pathology and the development of these diseases has highlighted novel regulators in the homeostatic maintenance of mitochondria. Importantly, this offers potential therapeutic targets in the development of future treatments for these degenerative diseases.
Related collections
Most cited references204
- Record: found
- Abstract: found
- Article: not found
Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis.
- Record: found
- Abstract: found
- Article: not found
Nix is a selective autophagy receptor for mitochondrial clearance.

- Record: found
- Abstract: found
- Article: found