- Record: found
- Abstract: found
- Article: found
Meiotic Drive Impacts Expression and Evolution of X-Linked Genes in Stalk-Eyed Flies
Read this article at
Abstract
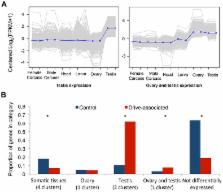
Although sex chromosome meiotic drive has been observed in a variety of species for over 50 years, the genes causing drive are only known in a few cases, and none of these cases cause distorted sex-ratios in nature. In stalk-eyed flies ( Teleopsis dalmanni), driving X chromosomes are commonly found at frequencies approaching 30% in the wild, but the genetic basis of drive has remained elusive due to reduced recombination between driving and non-driving X chromosomes. Here, we used RNAseq to identify transcripts that are differentially expressed between males carrying either a driving X (X SR) or a standard X chromosome (X ST), and found hundreds of these, the majority of which are X-linked. Drive-associated transcripts show increased levels of sequence divergence (dN/dS) compared to a control set, and are predominantly expressed either in testes or in the gonads of both sexes. Finally, we confirmed that X SR and X ST are highly divergent by estimating sequence differentiation between the RNAseq pools. We found that X-linked transcripts were often strongly differentiated (whereas most autosomal transcripts were not), supporting the presence of a relatively large region of recombination suppression on X SR presumably caused by one or more inversions. We have identified a group of genes that are good candidates for further study into the causes and consequences of sex-chromosome drive, and demonstrated that meiotic drive has had a profound effect on sequence evolution and gene expression of X-linked genes in this species.
Author Summary
Sex chromosome meiotic drive causes changes in the sex-ratios of natural populations, and may even lead to extinction if the driving element reaches high frequency. However, very little is known about the genes that cause sex-ratio drive, and no causal gene has been identified in a species that consistently exhibits distorted sex ratios in natural populations. Several species of stalk-eyed flies in southeast Asia – genus Teleopsis – express X chromosome drive, but the genes underlying drive have been difficult to locate due to reduced recombination between drive and standard X chromosomes presumably caused by the presence of a large inversion. Here, we use high throughput RNA sequencing to identify over 500 transcripts that are differentially expressed in the testes due to the effects of a driving X chromosome (X SR) in T. dalmanni. Most of these are X-linked, evolve more rapidly than control genes, and exhibit elevated expression in the gonads. Finally, X SR has become genetically differentiated from standard X chromosomes – using the RNA sequence data, we found nearly 1000 sites in X-linked transcripts and only a handful in autosomal transcripts where there was a fixed nucleotide difference. We conclude that X SR has led to widespread sequence and expression divergence on the X chromosome in T. dalmanni.
Related collections
Most cited references53
- Record: found
- Abstract: found
- Article: not found
Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material.
- Record: found
- Abstract: not found
- Article: not found
THE RELATION OF RECOMBINATION TO MUTATIONAL ADVANCE.

- Record: found
- Abstract: found
- Article: found