- Record: found
- Abstract: found
- Article: found
Analysis of the blood bacterial composition of patients with acute coronary syndrome and chronic coronary syndrome
Read this article at
Abstract
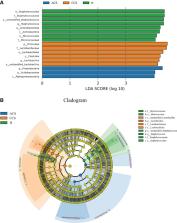
Emerging evidence revealed that the blood microbiota plays a role in several non-communicable diseases, including cardiovascular disease. However, the role of circulating microbes in atherosclerosis remains understudied. To test this hypothesis, we performed this study to investigate the microbial profile in the blood of Chines atherosclerosis volunteers. A total of seventy Acute Coronary Syndrome patients, seventy Chronic Coronary Syndrome patients, and seventy healthy individuals were examined using high-throughput Illumina Novaseq targeting the V3-V4 regions of the 16S rRNA gene. The relationship between atherosclerosis and blood microbiome, clinical variables, and their functional pathways were also investigated. Our study observed significantly higher alpha diversity indices (Chao1, p = 0.001, and Shannon, p = 0.004) in the acute coronary syndrome group compared with chronic coronary syndrome and healthy group, although a significantly lower alpha diversity was observed in the chronic coronary syndrome compared to acute coronary syndrome and healthy group. Beta diversity based on principal coordinate analysis demonstrated a major separation among the three groups. In addition, using linear discriminant analysis, a significant distinct taxon such as Actinobacteria _ phylum, and Staphylococcus_ genus in the healthy group; Firmicutes_ phylum, and Lactobacillus_ genus in the chronic coronary syndrome group, and Proteobacteria and Acidobacteriota _ phyla in acute coronary syndrome group were observed among three groups. Clusters of Orthologous Genes grouped and Kyoto Encyclopedia of Genes and Genomes pathways suggested a significant variation among all groups ( p < 0.05). The blood microbiota analysis provides potential biomarkers for the detection of coronary syndromes in this population.
Related collections
Most cited references75
- Record: found
- Abstract: found
- Article: not found
Heart Disease and Stroke Statistics—2016 Update
- Record: found
- Abstract: found
- Article: not found
Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities.
- Record: found
- Abstract: found
- Article: not found