- Record: found
- Abstract: found
- Article: not found
Genomic profiling of a randomized trial of interferon-α vs hydroxyurea in MPN reveals mutation-specific responses


Key Points
Visual Abstract
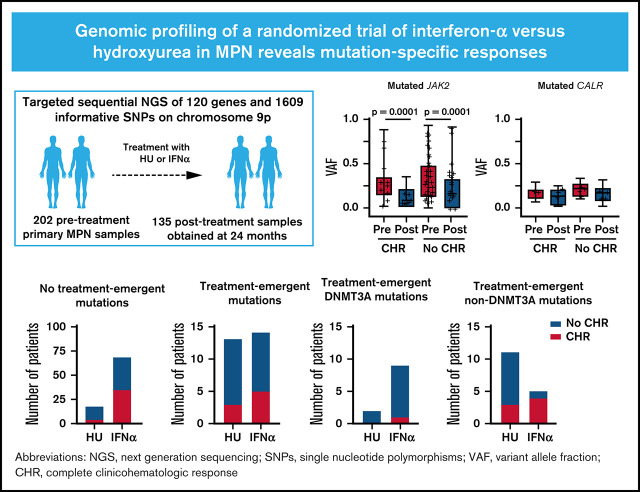
Abstract
Although somatic mutations influence the pathogenesis, phenotype, and outcome of myeloproliferative neoplasms (MPNs), little is known about their impact on molecular response to cytoreductive treatment. We performed targeted next-generation sequencing (NGS) on 202 pretreatment samples obtained from patients with MPN enrolled in the DALIAH trial (A Study of Low Dose Interferon Alpha Versus Hydroxyurea in Treatment of Chronic Myeloid Neoplasms; #NCT01387763), a randomized controlled phase 3 clinical trial, and 135 samples obtained after 24 months of therapy with recombinant interferon-alpha (IFNα) or hydroxyurea. The primary aim was to evaluate the association between complete clinicohematologic response (CHR) at 24 months and molecular response through sequential assessment of 120 genes using NGS. Among JAK2-mutated patients treated with IFNα, those with CHR had a greater reduction in the JAK2 variant allele frequency (median, 0.29 to 0.07; P < .0001) compared with those not achieving CHR (median, 0.27 to 0.14; P < .0001). In contrast, the CALR variant allele frequency did not significantly decline in those achieving CHR or in those not achieving CHR. Treatment-emergent mutations in DNMT3A were observed more commonly in patients treated with IFNα compared with hydroxyurea ( P = .04). Furthermore, treatment-emergent DNMT3A mutations were significantly enriched in IFNα–treated patients not attaining CHR ( P = .02). A mutation in TET2, DNMT3A, or ASXL1 was significantly associated with prior stroke (age-adjusted odds ratio, 5.29; 95% confidence interval, 1.59-17.54; P = .007), as was a mutation in TET2 alone (age-adjusted odds ratio, 3.03; 95% confidence interval, 1.03-9.01; P = .044). At 24 months, we found mutation-specific response patterns to IFNα: (1) JAK2- and CALR-mutated MPN exhibited distinct molecular responses; and (2) DNMT3A-mutated clones/subclones emerged on treatment.
Related collections
Most cited references70
- Record: found
- Abstract: found
- Article: not found
The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia.
- Record: found
- Abstract: found
- Article: not found
Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease
- Record: found
- Abstract: found
- Article: not found