- Record: found
- Abstract: found
- Article: found
A spontaneous complex structural variant in rcan-1 increases exploratory behavior and laboratory fitness of Caenorhabditis elegans
Read this article at
Abstract
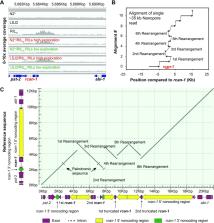
Over long evolutionary timescales, major changes to the copy number, function, and genomic organization of genes occur, however, our understanding of the individual mutational events responsible for these changes is lacking. In this report, we study the genetic basis of adaptation of two strains of C. elegans to laboratory food sources using competition experiments on a panel of 89 recombinant inbred lines (RIL). Unexpectedly, we identified a single RIL with higher relative fitness than either of the parental strains. This strain also displayed a novel behavioral phenotype, resulting in higher propensity to explore bacterial lawns. Using bulk-segregant analysis and short-read resequencing of this RIL, we mapped the change in exploration behavior to a spontaneous, complex rearrangement of the rcan-1 gene that occurred during construction of the RIL panel. We resolved this rearrangement into five unique tandem inversion/duplications using Oxford Nanopore long-read sequencing. rcan-1 encodes an ortholog to human RCAN1/DSCR1 calcipressin gene, which has been implicated as a causal gene for Down syndrome. The genomic rearrangement in rcan-1 creates two complete and two truncated versions of the rcan-1 coding region, with a variety of modified 5’ and 3’ non-coding regions. While most copy-number variations (CNVs) are thought to act by increasing expression of duplicated genes, these changes to rcan-1 ultimately result in the reduction of its whole-body expression due to changes in the upstream regions. By backcrossing this rearrangement into a common genetic background to create a near isogenic line (NIL), we demonstrate that both the competitive advantage and exploration behavioral changes are linked to this complex genetic variant. This NIL strain does not phenocopy a strain containing an rcan-1 loss-of-function allele, which suggests that the residual expression of rcan-1 is necessary for its fitness effects. Our results demonstrate how colonization of new environments, such as those encountered in the laboratory, can create evolutionary pressure to modify gene function. This evolutionary mismatch can be resolved by an unexpectedly complex genetic change that simultaneously duplicates and diversifies a gene into two uniquely regulated genes. Our work shows how complex rearrangements can act to modify gene expression in ways besides increased gene dosage.
Author summary
Evolution acts on genetic variants that modify phenotypes that increase the likelihood of staying alive and passing on these genetic changes to subsequent generations (i.e. fitness). There is general interest in understanding the types of genetic variants that can increase fitness in specific environments. One route that fitness can be increased is through changes in behavior, such as finding new food sources. Here, we identify a spontaneous genetic change that increases exploration behavior and fitness of animals in laboratory environments. Interestingly, this genetic change is not a simple genetic change that deletes or changes the sequence of a protein product, but rather a complex structural variant that simultaneously duplicates the rcan-1 gene and also modifies its expression in a number of tissues. Our work demonstrates how a complex structural change can duplicate a gene, modify the DNA control regions that determine its cellular sites of action, and confer a fitness advantage that could lead to its spread in a population.
Related collections
Most cited references60
- Record: found
- Abstract: found
- Article: not found
Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project.

- Record: found
- Abstract: found
- Article: found
SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data
- Record: found
- Abstract: found
- Article: not found