- Record: found
- Abstract: found
- Article: found
Case Report: Hypomorphic Function and Somatic Reversion in DOCK8 Deficiency in One Patient With Two Novel Variants and Sclerosing Cholangitis
Read this article at
Abstract
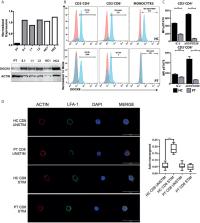
DOCK8 deficiency is a combined immunodeficiency due to biallelic variants in dedicator of cytokinesis 8 ( DOCK8) gene. The disease has a wide clinical spectrum encompassing recurrent infections (candidiasis, viral and bacterial infections), virally driven malignancies and immune dysregulatory features, including autoimmune (cytopenia and vasculitis) as well as allergic disorders (eczema, asthma, and food allergy). Hypomorphic function and somatic reversion of DOCK8 has been reported to result in incomplete phenotype without IgE overproduction. Here we describe a case of DOCK8 deficiency in a 8-year-old Caucasian girl. The patient’s disease was initially classified as autoimmune thrombocytopenia, which then evolved toward a combined immunodeficiency phenotype with recurrent infections, persistent EBV infection and lymphoproliferation. Two novel variants (one deletion and one premature stop codon) were characterized, resulting in markedly reduced, but not absent, DOCK8 expression. Somatic reversion of the DOCK8 deletion was identified in T cells. Hypomorphic function and somatic reversion were associated with restricted T cell repertoire, decreased STAT5 phosphorylation and impaired immune synapse functioning in T cells. Although the patient presented with incomplete phenotype (absence of markedly increase IgE and eosinophil count), sclerosing cholangitis was incidentally detected, thus indicating that hypomorphic function and somatic reversion of DOCK8 may delay disease progression but do not necessarily prevent from severe complications.
Related collections
Most cited references23
- Record: found
- Abstract: found
- Article: not found
Combined immunodeficiency associated with DOCK8 mutations.
- Record: found
- Abstract: found
- Article: not found
Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome.
- Record: found
- Abstract: found
- Article: found