- Record: found
- Abstract: found
- Article: found
Sequential deregulation of histone marks, chromatin accessibility and gene expression in response to PROTAC-induced degradation of ASH2L
Read this article at
Abstract
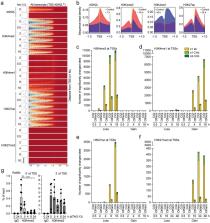
The trithorax protein ASH2L is essential for organismal and tissue development. As a subunit of COMPASS/KMT2 complexes, ASH2L is necessary for methylation of histone H3 lysine 4 (H3K4). Mono- and tri-methylation at this site mark active enhancers and promoters, respectively, although the functional relevance of H3K4 methylation is only partially understood. ASH2L has a long half-life, which results in a slow decrease upon knockout. This has made it difficult to define direct consequences. To overcome this limitation, we employed a PROTAC system to rapidly degrade ASH2L and address direct effects. ASH2L loss resulted in inhibition of proliferation of mouse embryo fibroblasts. Shortly after ASH2L degradation H3K4me3 decreased with its half-life varying between promoters. Subsequently, H3K4me1 increased at promoters and decreased at some enhancers. H3K27ac and H3K27me3, histone marks closely linked to H3K4 methylation, were affected with considerable delay. In parallel, chromatin compaction increased at promoters. Of note, nascent gene transcription was not affected early but overall RNA expression was deregulated late after ASH2L loss. Together, these findings suggest that downstream effects are ordered but relatively slow, despite the rapid loss of ASH2L and inactivation of KMT2 complexes. It appears that the systems that control gene transcription are well buffered and strong effects are only beginning to unfold after considerable delay.
Related collections
Most cited references93

- Record: found
- Abstract: found
- Article: found
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools
- Record: found
- Abstract: found
- Article: not found