- Record: found
- Abstract: found
- Article: found
Massive expansion of human gut bacteriophage diversity

Read this article at
Summary
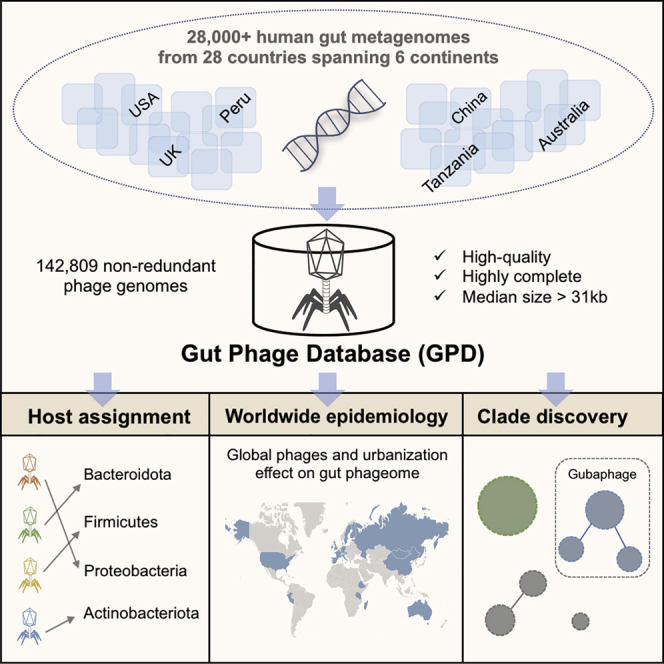
Bacteriophages drive evolutionary change in bacterial communities by creating gene flow networks that fuel ecological adaptions. However, the extent of viral diversity and its prevalence in the human gut remains largely unknown. Here, we introduce the Gut Phage Database, a collection of ∼142,000 non-redundant viral genomes (>10 kb) obtained by mining a dataset of 28,060 globally distributed human gut metagenomes and 2,898 reference genomes of cultured gut bacteria. Host assignment revealed that viral diversity is highest in the Firmicutes phyla and that ∼36% of viral clusters (VCs) are not restricted to a single species, creating gene flow networks across phylogenetically distinct bacterial species. Epidemiological analysis uncovered 280 globally distributed VCs found in at least 5 continents and a highly prevalent phage clade with features reminiscent of p-crAssphage. This high-quality, large-scale catalog of phage genomes will improve future virome studies and enable ecological and evolutionary analysis of human gut bacteriophages.
Graphical Abstract
Highlights
-
•
Database containing 142,809 non-redundant gut phage genomes from 28,060 metagenomes
-
•
Host assignment reveals phage diversity and host range across gut bacteria isolates
-
•
Epidemiology analysis unveils 280 viral clusters that are globally distributed
-
•
The Gubaphage is a clade that infects several members of the Bacteroidales order
Abstract
By mining human gut metagenomes and gut bacteria isolates, Camarillo-Guerrero et al. compile high-quality gut bacteriophage genomes into the Gut Phage Database (GPD) and analyze the diversity and worldwide distribution of phage.
Related collections
Most cited references73

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools

- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform
- Record: found
- Abstract: found
- Article: not found