- Record: found
- Abstract: found
- Article: found
OSR1 disruption contributes to uterine factor infertility via impaired Müllerian duct development and endometrial receptivity

Read this article at
Abstract
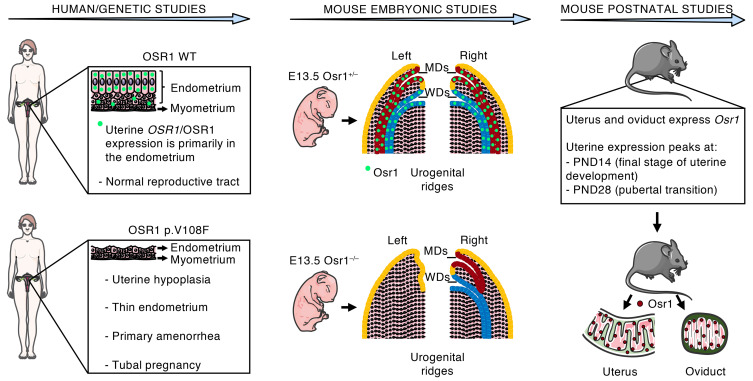
Three sisters, born from consanguineous parents, manifested a unique Müllerian anomaly characterized by uterine hypoplasia with thin estrogen-unresponsive endometrium and primary amenorrhea, but with spontaneous tubal pregnancies. Through whole-exome sequencing followed by comprehensive genetic analysis, a missense variant was identified in the OSR1 gene. We therefore investigated OSR1/OSR1 expression in postpubertal human uteri, and the prenatal and postnatal expression pattern of Osr1/Osr1 in murine developing Müllerian ducts (MDs) and endometrium, respectively. We then investigated whether Osr1 deletion would affect MD development, using WT and genetically engineered mice. Human uterine OSR1/OSR1 expression was found primarily in the endometrium. Mouse Osr1 was expressed prenatally in MDs and Wolffian ducts (WDs), from rostral to caudal segments, in E13.5 embryos. MDs and WDs were absent on the left side and MDs were rostrally truncated on the right side of E13.5 Osr1 –/– embryos. Postnatally, Osr1 was expressed in mouse uteri throughout their lifespan, peaking at postnatal days 14 and 28. Osr1 protein was present primarily in uterine luminal and glandular epithelial cells and in the epithelial cells of mouse oviducts. Through this translational approach, we demonstrated that OSR1 in humans and mice is important for MD development and endometrial receptivity and may be implicated in uterine factor infertility.
Abstract
Related collections
Most cited references42
- Record: found
- Abstract: found
- Article: not found
Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method.

- Record: found
- Abstract: found
- Article: found
The mutational constraint spectrum quantified from variation in 141,456 humans
- Record: found
- Abstract: found
- Article: not found