- Record: found
- Abstract: found
- Article: found
Fine-mapping across diverse ancestries drives the discovery of putative causal variants underlying human complex traits and diseases
Read this article at
Abstract
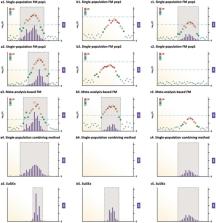
Genome-wide association studies (GWAS) of human complex traits or diseases often implicate genetic loci that span hundreds or thousands of genetic variants, many of which have similar statistical significance. While statistical fine-mapping in individuals of European ancestries has made important discoveries, cross-population fine-mapping has the potential to improve power and resolution by capitalizing on the genomic diversity across ancestries. Here we present SuSiEx, an accurate and computationally efficient method for cross-population fine-mapping, which builds on the single-population fine-mapping framework, Sum of Single Effects (SuSiE). SuSiEx integrates data from an arbitrary number of ancestries, explicitly models population-specific allele frequencies and LD patterns, accounts for multiple causal variants in a genomic region, and can be applied to GWAS summary statistics. We comprehensively evaluated SuSiEx using simulations, a range of quantitative traits measured in both UK Biobank and Taiwan Biobank, and schizophrenia GWAS across East Asian and European ancestries. In all evaluations, SuSiEx fine-mapped more association signals, produced smaller credible sets and higher posterior inclusion probability (PIP) for putative causal variants, and captured population-specific causal variants.
Related collections
Most cited references45

- Record: found
- Abstract: found
- Article: found
A global reference for human genetic variation

- Record: found
- Abstract: found
- Article: found
Second-generation PLINK: rising to the challenge of larger and richer datasets

- Record: found
- Abstract: found
- Article: found