- Record: found
- Abstract: found
- Article: found
Fatal Prion Disease in a Mouse Model of Genetic E200K Creutzfeldt-Jakob Disease
Read this article at
Abstract
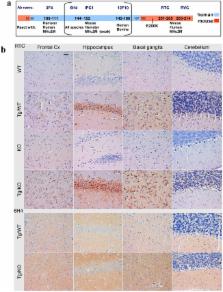
Genetic prion diseases are late onset fatal neurodegenerative disorders linked to pathogenic mutations in the prion protein-encoding gene, PRNP. The most prevalent of these is the substitution of Glutamate for Lysine at codon 200 (E200K), causing genetic Creutzfeldt-Jakob disease (gCJD) in several clusters, including Jews of Libyan origin. Investigating the pathogenesis of genetic CJD, as well as developing prophylactic treatments for young asymptomatic carriers of this and other PrP mutations, may well depend upon the availability of appropriate animal models in which long term treatments can be evaluated for efficacy and toxicity. Here we present the first effective mouse model for E200KCJD, which expresses chimeric mouse/human (TgMHu2M) E199KPrP on both a null and a wt PrP background, as is the case for heterozygous patients and carriers. Mice from both lines suffered from distinct neurological symptoms as early as 5–6 month of age and deteriorated to death several months thereafter. Histopathological examination of the brain and spinal cord revealed early gliosis and age-related intraneuronal deposition of disease-associated PrP similarly to human E200K gCJD. Concomitantly we detected aggregated, proteinase K resistant, truncated and oxidized PrP forms on immunoblots. Inoculation of brain extracts from TgMHu2ME199K mice readily induced, the first time for any mutant prion transgenic model, a distinct fatal prion disease in wt mice. We believe that these mice may serve as an ideal platform for the investigation of the pathogenesis of genetic prion disease and thus for the monitoring of anti-prion treatments.
Author Summary
Inherited prion diseases, such as genetic CJD, are dominant disorders linked to mutations in the gene encoding the prion protein, PrP. Since therapeutic intervention in all types of human prion diseases has failed, we propose that therapeutic efforts should be directed mostly to the development of preventive treatments for subjects incubating prion diseases, as is the case for asymptomatic carriers of pathogenic PrP mutations. These subjects will develop disease symptoms at some point in their adult life; therefore they should be treated before clinical deterioration. Candidate treatments will need to be tested for efficacy and safety first in animal models that mimic most properties of genetic CJD. In this work, we describe a new transgenic mouse model for E200K genetic CJD, presenting progressive neurodegenerative disease and age related prion disease pathology and biochemistry, as is the case in the human disease. Brain extracts from these mice also transmitted prion disease to wt mice, as shown before for parallel human samples. We propose that these animals will play a significant role in the development of novel anti-prion prophylactic treatments.
Related collections
Most cited references44
- Record: found
- Abstract: found
- Article: not found
Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein.
- Record: found
- Abstract: found
- Article: not found
Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease.
- Record: found
- Abstract: found
- Article: not found