- Record: found
- Abstract: found
- Article: found
scSNV: accurate dscRNA-seq SNV co-expression analysis using duplicate tag collapsing
Read this article at
Abstract
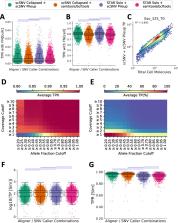
Identifying single nucleotide variants has become common practice for droplet-based single-cell RNA-seq experiments; however, presently, a pipeline does not exist to maximize variant calling accuracy. Furthermore, molecular duplicates generated in these experiments have not been utilized to optimally detect variant co-expression. Herein, we introduce scSNV designed from the ground up to “collapse” molecular duplicates and accurately identify variants and their co-expression. We demonstrate that scSNV is fast, with a reduced false-positive variant call rate, and enables the co-detection of genetic variants and A>G RNA edits across twenty-two samples.
Related collections
Most cited references31

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools
- Record: found
- Abstract: found
- Article: not found
STAR: ultrafast universal RNA-seq aligner.
- Record: found
- Abstract: found
- Article: not found