- Record: found
- Abstract: found
- Article: found
DUX4 double whammy: The transcription factor that causes a rare muscular dystrophy also kills the precursors of the human nose
Read this article at
Abstract
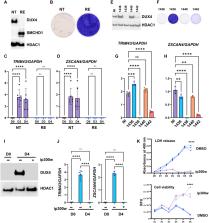
SMCHD1 mutations cause congenital arhinia (absent nose) and a muscular dystrophy called FSHD2. In FSHD2, loss of SMCHD1 repressive activity causes expression of double homeobox 4 (DUX4) in muscle tissue, where it is toxic. Studies of arhinia patients suggest a primary defect in nasal placode cells (human nose progenitors). Here, we show that upon SMCHD1 ablation, DUX4 becomes derepressed in H9 human embryonic stem cells (hESCs) as they differentiate toward a placode cell fate, triggering cell death. Arhinia and FSHD2 patient-derived induced pluripotent stem cells (iPSCs) express DUX4 when converted to placode cells and demonstrate variable degrees of cell death, suggesting an environmental disease modifier. HSV-1 may be one such modifier as herpesvirus infection amplifies DUX4 expression in SMCHD1 KO hESC and patient iPSC. These studies suggest that arhinia, like FSHD2, is due to compromised SMCHD1 repressive activity in a cell-specific context and provide evidence for an environmental modifier.
Abstract

Abstract
Loss of epigenetic silencing by the chromatin remodeler SMCHD1 unleashes the DUX4 muscle toxin which kills cranial placode cells.
Related collections
Most cited references73
- Record: found
- Abstract: found
- Article: not found
STAR: ultrafast universal RNA-seq aligner.
- Record: found
- Abstract: found
- Article: not found
Fast gapped-read alignment with Bowtie 2.
- Record: found
- Abstract: found
- Article: not found