- Record: found
- Abstract: found
- Article: found
Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy†
Read this article at
Abstract
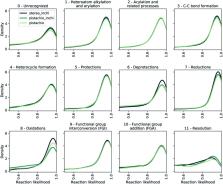
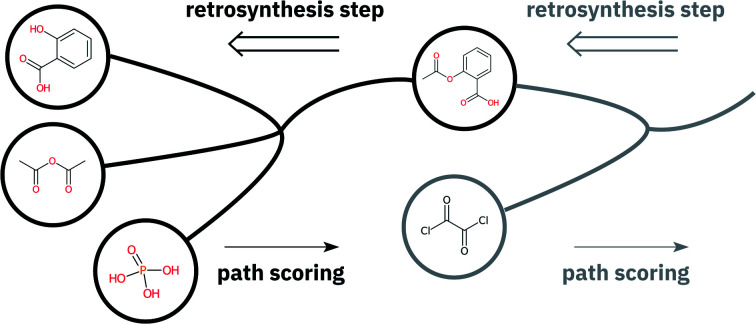
We present an extension of our Molecular Transformer model combined with a hyper-graph exploration strategy for automatic retrosynthesis route planning without human intervention. The single-step retrosynthetic model sets a new state of the art for predicting reactants as well as reagents, solvents and catalysts for each retrosynthetic step. We introduce four metrics (coverage, class diversity, round-trip accuracy and Jensen–Shannon divergence) to evaluate the single-step retrosynthetic models, using the forward prediction and a reaction classification model always based on the transformer architecture. The hypergraph is constructed on the fly, and the nodes are filtered and further expanded based on a Bayesian-like probability. We critically assessed the end-to-end framework with several retrosynthesis examples from literature and academic exams. Overall, the frameworks have an excellent performance with few weaknesses related to the training data. The use of the introduced metrics opens up the possibility to optimize entire retrosynthetic frameworks by focusing on the performance of the single-step model only.
Abstract
We present an extension of our Molecular Transformer model combined with a hyper-graph exploration strategy for automatic retrosynthesis route planning without human intervention.
Related collections
Most cited references26
- Record: found
- Abstract: found
- Article: not found
Planning chemical syntheses with deep neural networks and symbolic AI
- Record: found
- Abstract: found
- Article: not found
A robotic platform for flow synthesis of organic compounds informed by AI planning

- Record: found
- Abstract: found
- Article: found