- Record: found
- Abstract: found
- Article: found
Ehf and Fezf2 regulate late medullary thymic epithelial cell and thymic tuft cell development
Read this article at
Abstract
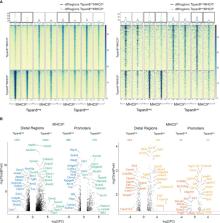
Thymic epithelial cells are indispensable for T cell maturation and selection and the induction of central immune tolerance. The self-peptide repertoire expressed by medullary thymic epithelial cells is in part regulated by the transcriptional regulator Aire (Autoimmune regulator) and the transcription factor Fezf2. Due to the high complexity of mTEC maturation stages (i.e., post-Aire, Krt10+ mTECs, and Dclk1+ Tuft mTECs) and the heterogeneity in their gene expression profiles (i.e., mosaic expression patterns), it has been challenging to identify the additional factors complementing the transcriptional regulation. We aimed to identify the transcriptional regulators involved in the regulation of mTEC development and self-peptide expression in an unbiased and genome-wide manner. We used ATAC footprinting analysis as an indirect approach to identify transcription factors involved in the gene expression regulation in mTECs, which we validated by ChIP sequencing. This study identifies Fezf2 as a regulator of the recently described thymic Tuft cells (i.e., Tuft mTECs). Furthermore, we identify that transcriptional regulators of the ELF, ESE, ERF, and PEA3 subfamily of the ETS transcription factor family and members of the Krüppel-like family of transcription factors play a role in the transcriptional regulation of genes involved in late mTEC development and promiscuous gene expression.
Related collections
Most cited references88

- Record: found
- Abstract: found
- Article: found
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2
- Record: found
- Abstract: found
- Article: not found
STAR: ultrafast universal RNA-seq aligner.
- Record: found
- Abstract: found
- Article: not found