- Record: found
- Abstract: found
- Article: found
A risk model based on pyroptosis subtypes predicts tumor immune microenvironment and guides chemotherapy and immunotherapy in bladder cancer
Read this article at
Abstract
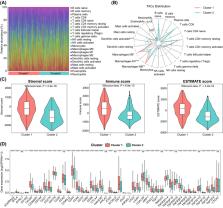
Although immunotherapy has revolutionized bladder cancer (BLCA) therapy, only few patients demonstrate durable clinical benefits due to the heterogeneity. Emerging evidence has linked pyroptosis to shaping tumor microenvironment (TME) and predicting therapy response. However, the relationship between pyroptosis and immunotherapy response in BLCA remains elusive. In this study, we performed a comprehensive bioinformatic analysis to dissect the role of pyroptosis in BLCA. Differentially expressed pyroptosis-related genes (DEPRGs) between tumor and normal tissues were identified using publicly available datasets. Kaplan–Meier analysis was performed to screen for DEPRGs associated with survival. Consensus clustering was used for BLCA subtyping. TME characteristics were evaluated by CIBERSORT, ESTIMATE and immune checkpoint genes (ICGs). Following univariate COX regression and LASSO analyses with pyroptosis-related DEGs, the risk model and nomogram were constructed with TCGA dataset and validated in the GEO dataset. Furthermore, therapeutic responses in high- and low-risk groups were compared using TIDE and GDSC databases. Two pyroptosis-related subtypes (Cluster 1 and 2) were identified based on expression patterns of GSDMA and CHMP4C. Bioinformatic analyses showed that cluster 1 had poor survival, more M0/M1/M2 macrophages, higher immune/stromal/ESTIMATE scores, and higher expression levels of ICGs. A 15-gene signature for predicting prognosis could classify patients into high- and low-risk groups. Furthermore, the correlation of risk scores with TIDE score and IC 50 showed that patients in low-risk group were more sensitive to immunotherapy, whereas patients in high-risk group could better benefit from chemotherapy. Our study identified two novel pyroptosis-related subtypes and constructed a risk model, which can predict the prognosis, improve our understanding the role of PRGs in BLCA, and guide chemotherapy and immunotherapy.
Related collections
Most cited references53
- Record: found
- Abstract: found
- Article: not found
Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries

- Record: found
- Abstract: found
- Article: found
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2

- Record: found
- Abstract: found
- Article: found