- Record: found
- Abstract: found
- Article: found
Different Sources of Allelic Variation Drove Repeated Color Pattern Divergence in Cichlid Fishes
Read this article at
Abstract
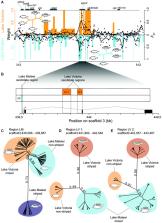
The adaptive radiations of East African cichlid fish in the Great Lakes Victoria, Malawi, and Tanganyika are well known for their diversity and repeatedly evolved phenotypes. Convergent evolution of melanic horizontal stripes has been linked to a single locus harboring the gene agouti-related peptide 2 ( agrp2). However, where and when the causal variants underlying this trait evolved and how they drove phenotypic divergence remained unknown. To test the alternative hypotheses of standing genetic variation versus de novo mutations (independently originating in each radiation), we searched for shared signals of genomic divergence at the agrp2 locus. Although we discovered similar signatures of differentiation at the locus level, the haplotypes associated with stripe patterns are surprisingly different. In Lake Malawi, the highest associated alleles are located within and close to the 5′ untranslated region of agrp2 and likely evolved through recent de novo mutations. In the younger Lake Victoria radiation, stripes are associated with two intronic regions overlapping with a previously reported cis-regulatory interval. The origin of these segregating haplotypes predates the Lake Victoria radiation because they are also found in more basal riverine and Lake Kivu species. This suggests that both segregating haplotypes were present as standing genetic variation at the onset of the Lake Victoria adaptive radiation with its more than 500 species and drove phenotypic divergence within the species flock. Therefore, both new (Lake Malawi) and ancient (Lake Victoria) allelic variation at the same locus fueled rapid and convergent phenotypic evolution.
Related collections
Most cited references94

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools

- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform

- Record: found
- Abstract: found
- Article: found