- Record: found
- Abstract: found
- Article: found
Multiple-trait QTL mapping and genomic prediction for wool traits in sheep
Read this article at
Abstract
Background
The application of genomic selection to sheep breeding could lead to substantial increases in profitability of wool production due to the availability of accurate breeding values from single nucleotide polymorphism (SNP) data. Several key traits determine the value of wool and influence a sheep’s susceptibility to fleece rot and fly strike. Our aim was to predict genomic estimated breeding values (GEBV) and to compare three methods of combining information across traits to map polymorphisms that affect these traits.
Methods
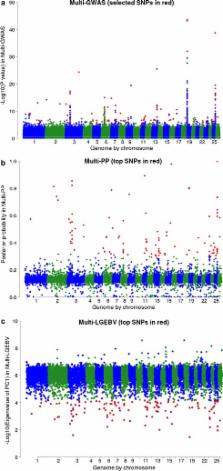
GEBV for 5726 Merino and Merino crossbred sheep were calculated using BayesR and genomic best linear unbiased prediction (GBLUP) with real and imputed 510,174 SNPs for 22 traits (at yearling and adult ages) including wool production and quality, and breech conformation traits that are associated with susceptibility to fly strike. Accuracies of these GEBV were assessed using fivefold cross-validation. We also devised and compared three approximate multi-trait analyses to map pleiotropic quantitative trait loci (QTL): a multi-trait genome-wide association study and two multi-trait methods that use the output from BayesR analyses. One BayesR method used local GEBV for each trait, while the other used the posterior probabilities that a SNP had an effect on each trait.
Results
BayesR and GBLUP resulted in similar average GEBV accuracies across traits (~0.22). BayesR accuracies were highest for wool yield and fibre diameter (>0.40) and lowest for skin quality and dag score (<0.10). Generally, accuracy was higher for traits with larger reference populations and higher heritability. In total, the three multi-trait analyses identified 206 putative QTL, of which 20 were common to the three analyses. The two BayesR multi-trait approaches mapped QTL in a more defined manner than the multi-trait GWAS. We identified genes with known effects on hair growth (i.e. FGF5, STAT3, KRT86, and ALX4) near SNPs with pleiotropic effects on wool traits.
Related collections
Most cited references57

- Record: found
- Abstract: found
- Article: found
A new approach for efficient genotype imputation using information from relatives
- Record: found
- Abstract: found
- Article: not found
Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels.

- Record: found
- Abstract: found
- Article: found
Simultaneous Discovery, Estimation and Prediction Analysis of Complex Traits Using a Bayesian Mixture Model
Author and article information
Comments
Comment on this article
See how this article has been cited at scite.ai
scite shows how a scientific paper has been cited by providing the context of the citation, a classification describing whether it supports, mentions, or contrasts the cited claim, and a label indicating in which section the citation was made.