- Record: found
- Abstract: found
- Article: found
Sensitization of human cancer cells to gemcitabine by the Chk1 inhibitor MK-8776: cell cycle perturbation and impact of administration schedule in vitro and in vivo
Read this article at
Abstract
Background
Chk1 inhibitors have emerged as promising anticancer therapeutic agents particularly when combined with antimetabolites such as gemcitabine, cytarabine or hydroxyurea. Here, we address the importance of appropriate drug scheduling when gemcitabine is combined with the Chk1 inhibitor MK-8776, and the mechanisms involved in the schedule dependence.
Methods
Growth inhibition induced by gemcitabine plus MK-8776 was assessed across multiple cancer cell lines. Experiments used clinically relevant “bolus” administration of both drugs rather than continuous drug exposures. We assessed the effect of different treatment schedules on cell cycle perturbation and tumor cell growth in vitro and in xenograft tumor models.
Results
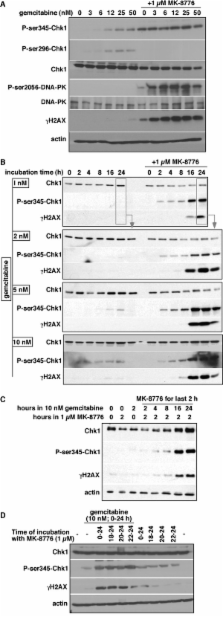
MK-8776 induced an average 7-fold sensitization to gemcitabine in 16 cancer cell lines. The time of MK-8776 administration significantly affected the response of tumor cells to gemcitabine. Although gemcitabine induced rapid cell cycle arrest, the stalled replication forks were not initially dependent on Chk1 for stability. By 18 h, RAD51 was loaded onto DNA indicative of homologous recombination. Inhibition of Chk1 at 18 h rapidly dissociated RAD51 leading to the collapse of replication forks and cell death. Addition of MK-8776 from 18–24 h after a 6-h incubation with gemcitabine induced much greater sensitization than if the two drugs were incubated concurrently for 6 h. The ability of this short incubation with MK-8776 to sensitize cells is critical because of the short half-life of MK-8776 in patients’ plasma. Cell cycle perturbation was also assessed in human pancreas tumor xenografts in mice. There was a dramatic accumulation of cells in S/G 2 phase 18 h after gemcitabine administration, but cells had started to recover by 42 h. Administration of MK-8776 18 h after gemcitabine caused significantly delayed tumor growth compared to either drug alone, or when the two drugs were administered with only a 30 min interval.
Conclusions
There are two reasons why delayed addition of MK-8776 enhances sensitivity to gemcitabine: first, there is an increased number of cells arrested in S phase; and second, the arrested cells have adequate time to initiate recombination and thereby become Chk1 dependent. These results have important implications for the design of clinical trials using this drug combination.
Related collections
Most cited references26
- Record: found
- Abstract: found
- Article: not found
Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening.
- Record: found
- Abstract: found
- Article: not found
Targeting the S and G2 checkpoint to treat cancer.
- Record: found
- Abstract: found
- Article: not found