- Record: found
- Abstract: found
- Article: found
Candida–streptococcal interactions in biofilm-associated oral diseases
review-article
13 December 2018
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Bacterial–fungal interactions and oral diseases
The oral cavity contains up to 700 different species of microorganisms, including
both bacteria and fungi [1]. The interactions of these communities of different organisms
has become of increasing interest, particularly with respect to cross-kingdom interactions
involving fungi and bacteria, which have been associated with severity of dental caries
(tooth decay) and mucosal infections. Here, we provide a short review of the significance
and mechanisms for the interactions between C. albicans and streptococci, the most
common fungal and bacterial organisms in the oral cavity [2–5].
Candida albicans and oral streptococci coinfections are associated with enhanced virulence
of dental caries and more severe oropharyngeal diseases (Fig 1) [6,7]. Specifically,
C. albicans partners with Streptococcus gordonii, S. oralis, and S. sanguinis to enhance
bacterial colonization and biofilm formation. In addition, C. albicans becomes more
invasive, exacerbating mucosal tissue infection and destruction [8,9]. Mixed C. albicans–bacterial
infections are also associated with denture stomatitis, the inflammation of the oral
mucosa under dentures. Furthermore, C. albicans–bacterial communities have been clinically
found in other oral niches, including periodontal pockets and endodontic canals [6].
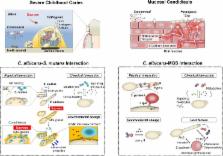
10.1371/journal.ppat.1007342.g001
Fig 1
Candida–streptococcal interactions and oral diseases.
A. Confocal fluorescence microscopy images of C. albicans–S. mutans mixed biofilms,
illustrating the spatial relationship between C. albicans (blue), S. mutans (green),
and exopolysaccharides (red). B. Images of teeth from rats infected with S. mutans,
C. albicans, or coinfected. Black arrows indicate severe carious lesions of coinfections
in which enamel is missing, which exposes underlying dentin. Such rampant caries was
absent in the animals infected by S. mutans or C. albicans alone. C. Fluorescence
microscopy images of harvested mouse tongues infected with S. oralis (red, see arrows),
C. albicans (green), or both. Coinfection substantially increased bacterial–fungal
biofilm accumulation, soft tissue invasion, and inflammatory response. Original images
provided by Dr. Anna Dongari-Bagtzoglou; adapted from Sobue T. and colleagues, Methods
Mol Biol. 1356:137–52, 2016, with permission. EPS, exopolysaccharides.
C. albicans–streptococcal biofilms are an important contributor to the development
of early childhood caries that affects toddler-age children [10–12]. Severe childhood
caries is a particularly virulent form of caries that causes extensive and painful
tooth destruction, induced by protracted consumption of sucrose containing foods and
beverages [11]. Typically, C. albicans is usually absent on teeth of healthy, caries-free
children [10]. Furthermore, C. albicans does not interact strongly with S. mutans
(a caries-causing pathogen), nor is it an efficient colonizer of mineralized tooth
enamel by itself. However, the high level of sucrose in the oral cavity increases
the physical coadhesion between the C. albicans and S. mutans as well as tooth surface
colonization and drastically enhances the microbial burden, aciduricity, and production
of extracellular matrix. Ultimately, the extensive mixed-kingdom and acidogenic biofilm
leads to severe tooth decay in a process that can be recapitulated in a rodent model
under sugar-rich diets [12].
Fungal and bacterial cell surface adhesins mediate C. albicans interactions with mitis
group streptococci on mucosal surfaces
C. albicans physically interacts with mitis group streptococci (MGS) species such
as S. gordonii, S. sanguinis, and S. oralis through well-characterized cell wall surface
proteins/receptors on both organisms [6,13]. Streptococcal cell surface adhesins SspA
and SspB (from the antigen I/II polypeptide family) interact with the C. albicans
surface, while ALS and HWP1 adhesins on the fungal cell wall appear to mediate binding
to MGS (Fig 2). Specifically, SspB and Als3 directly bind C. albicans and S. gordonii
together through the N-terminal domain of Als3 [14]. These interactions may also involve
O-mannosyl residues in Als adhesins and other cell wall proteins, such as Sap9 [15,16].
10.1371/journal.ppat.1007342.g002
Fig 2
Pathogenic mechanisms of C. albicans–oral streptococcal cross-kingdom interactions.
Complex physical and chemical interactions (including cross-feeding and metabolites
exchange) as well as environmental and host factors govern the development of pathogenic
bacterial–fungal biofilms, including spatial organization, virulence, and drug protection/resistance.
These interactions can be cooperative or competitive to mediate symbiotic, antagonistic,
or synergistic relationships, often modulated by host and environmental factors to
promote the onset and amplify the severity of the disease. Host diet (dietary sugars,
particularly sucrose) promote the interactions between C. albicans and S. mutans by
providing a substrate for EPS α-glucans production by streptococcal Gtfs that enhances
coadhesion and bacterial–fungal tooth colonization, stimulating cross-kingdom biofilms.
This interaction enhances the carriage of the cariogenic pathogen and acid production,
while the presence of C. albicans increases EPS matrix production (via Gtf induction
and fungal-derived EPS) and biofilm aciduricity, resulting in cariogenic conditions
on tooth surface. Likewise, the pathogenic impact of C. albicans interactions with
MGS on mucosal surfaces is also influenced by host factors. The interactions of S.
oralis with C. albicans on mucosal surfaces cause exacerbated inflammatory responses
and increased neutrophilic activity. C. albicans increase the biomass of S. oralis
and this leads to increased mucosal TLR2 expression, activating proinflammatory signaling.
C. albicans and S. oralis also synergistically increase epithelial μ-calpain activity,
a proteolytic enzyme that targets E-cadherin from epithelial junctions. The bacteria
influence fungal physiology by promoting hyphal formation via the Efg1 filamentation
pathway and expression of secreted aspartyl proteases, which further induces proteolytic
degradation of E-cadherin, facilitating invasion and tissue destruction. Efg1; EPS,
exopolysaccharides; Gtf, glucosyltransferase; MGS, mitis group streptococci; TLR2.
The consequences of C. albicans–streptococcal interactions have been demonstrated
in vivo. C. albicans and S. oralis coinfection results in increased tissue invasion
and heightened mucosal inflammatory responses compared with infection by either organism
alone [9]. This latter feature appears to be due to increased induction of multiple
neutrophil-activating cytokines and up-regulation of TLR2-dependent inflammatory genes
as well as enhanced epithelial μ-calpain activity [9,17] (Fig 2).
C. albicans interacts with S. mutans exoenzymes (glucosyltransferases) to promote
interspecies biofilm formation on tooth surface
In contrast to MGS, C. albicans does not directly bind to the cariogenic pathogen
S. mutans. Instead, glucosyltransferases (Gtfs) secreted by S. mutans promote the
generation of an extensive extracellular matrix in the presence of C. albicans, leading
to virulent mixed biofilms under sugar-rich conditions of severe childhood caries
[18,19]. S. mutans-derived GtfB binds avidly to the C. albicans cell surface and converts
sucrose to large amounts of extracellular polysaccharides (EPS) α-glucans on the fungal
surface (Fig 2). The EPS provides bacterial binding sites for S. mutans and concurrently
allows C. albicans to bind to and colonize teeth [12]. Consequently, the interaction
between S. mutans and C. albicans is mediated by both secreted Gtfs and their glucan
product. This mechanism is distinct from the more typical cell–cell binding interactions
observed between MGS, staphylococci, or bacillus and C. albicans, although the role
of Gtfs in MGS species has not been extensively studied [13].
To further understand the mechanistic basis of this “biochemical interaction,” we
identified the C. albicans surface molecules to which GtfB binds. C. albicans mutants
lacking either N- or O-linked mannans (located on the outer most layer of the fungal
cell wall) showed severely reduced GtfB binding. As a result, these mannoprotein-deficient
mutants developed poor mixed-species biofilms with S. mutans, showed reduced EPS α-glucans
content, and reduced microbial carriage on teeth in vivo [18]. Likewise, S. mutans
defective in GtfB does not yield mixed-species biofilms with C. albicans.
In a rodent model, the sucrose-dependent partnership between C. albicans and S. mutans
synergistically enhanced bacterial–fungal carriage within plaque biofilm, leading
to aggressive onset of tooth decay with rampant carious lesions similar to those found
in severe childhood caries [12]. The potential mechanisms for severe caries have been
an active subject of research, which entails at least, in part, enhanced microbial
carriage, cross-feeding metabolic interactions, and the accumulation of adherent acidic
biofilms on teeth facilitated by an EPS matrix surrounding acidogenic–aciduric organisms,
as reviewed recently [5,10] (Fig 2).
The role of the C. albicans master regulator Efg1 is required for MGS mixed biofilms
but not for S. mutans mixed biofilms
C. albicans Efg1 regulates key hyphae-associated biofilm effector molecules, and homozygous
efg1 deletion mutants form only rudimentary biofilms [20]. The nature of the biofilm
formed between S. mutans and C. albicans differs from single species C. albicans biofilms
because deletion of two transcription factors that are essential for C. albicans biofilms,
Efg1 and Bcr1, does not affect the amount of fungal cells in the mixed biofilm. This
is likely due to the fact that GtfB binds these mutants with similar afinity compared
to wild types and generates robust extracellular α-glucans matrix that allows C. albicans
to coadhere and form biofilm with S. mutans [18].
In contrast, efg1ΔΔ mutants are unable to form mixed biofilms with S. oralis [21].
Interestingly, overexpression of the Efg1-regulated adhesin ALS1 partially restores
C. albicans–S. oralis biofilm formation to efg1ΔΔ, suggesting that Als1 is a key mediator
of this mixed biofilm [21]. Consistent with this notion, C. albicans strains lacking
either ALS1 or ALS3 also are deficient for S. oralis mixed biofilm formation [21].
Als3 is also crucial for C. albicans–S. gordonii mixed biofilms through a mechanism
involving an interaction between Als3 and SspB [22]. However, C. albicans strains
lacking ALS3 are able to form mixed biofilms with S. mutans under sucrose-rich conditions,
showing similar levels of fungal cells as those formed with wild-type strains [18].
Thus, the interactions of C. albicans with oral streptococci vary significantly with
the specific species of bacteria. Additional studies will be needed to understand
how these differences affect the colonization and disease severity at distinct oral
niches.
C. albicans–bacterial biofilm relationship is critically dependent on EPS matrix and
chemical interactions
The EPS matrix critically influences the relationship between C. albicans and oral
streptococci within the biofilm [23]. The matrix provides a scaffold for both surface
adhesion and cell-to-cell cohesion while at the same time establishing chemical and
nutrient gradients by modulating diffusion [5]. Like most microbes, the matrix of
Candida species is comprised of the protein, carbohydrate, nucleic acid, and lipids.
In particular, a complex containing mannan and β-glucan constituents sequesters antifungal
drugs to protect Candida cells from their effects [23]. Nearly a dozen C. albicans
proteins involved in polysaccharide synthesis and modification (e.g., Phr1, Bgl2,
Alg11, and Mnn11) are indispensable for production of the matrix [23]. In mixed biofilms,
the fungal derived biofilm matrix also protects some prokaryotic pathogens (e.g.,
Staphylococcus aureus and Escherichia coli) against antibacterial drugs [24]. Similarly,
S. mutans-derived α-glucans surrounding fungal cells form an additional “drug-trapping
matrix” that prevents uptake of the antifungal fluconazole, reducing killing efficacy
[25].
Complex signaling, cross-feeding, and metabolic interactions within the biofilm shape
its microenvironment and lead to pathogenic synergies that modulate the onset and
severity of oral diseases (Fig 2). A range of signaling/quorum sensing (QS) molecules
and other factors appear to facilitate these synergies, including AI-2, peptidoglycan
fragments, exoenzymes, and hydrogen peroxide (H2O2) [2–4,13,26]. For example, nutrient
byproducts as well as AI-2 signaling and H2O2 from S. gordonii stimulate C. albicans
hyphal development within the biofilm [26], while S. oralis presence also activates
expression of fungal aspartyl proteases [13]. Conversely, C. albicans can promote
streptococcal proliferation by providing growth-stimulating factors and reducing oxygen
tension [13,26]. The impact of C. albicans and MGS synergism on the host–pathogen
interaction has been demonstrated in vivo whereby mixed biofilm (with S. oralis) growth
enhances neutrophil infiltration, leading to increased severity of soft tissue lesions
[9,17] (Fig 2). This is distinct from single-species C. albicans biofilms, which are
notable for inhibition of neutrophil influx and subsequent function.
A further example of the consequences of the EPS matrix and chemical interactions
has been observed between C. albicans and S. mutans. S. mutans converts sucrose to
glucose that can be more readily metabolized by C. albicans [27,28, 29]. Importantly,
C. albicans activates S. mutans competence [28], virulence genes, and GtfB production
via QS molecules such as farnesol [27]. Furthermore, C. albicans secretes its own
matrix products such as β-glucan and creates an EPS-producing loop within the S. mutans
mixed biofilm [12]. As a result, the organisms enhance the carriage of cariogenic
pathogens, biofilm accumulation, and acid production, promoting a localized and persistent
acidogenic–aciduric microenvironment that potentiates demineralization of tooth enamel
and may explain the synergistic enhancement of caries severity.
Although the cross-kingdom synergies are involved in the pathogenesis of both mucosal
and dental diseases, the interactions can also repress functions of the member species
to modulate population growth, biofilm structure, community changes, and spatial organization
[5] (Fig 2). For example, S. mutans-derived metabolites such as mutanobactin A and
fatty acid signaling trans-2-decenoic acid inhibit C. albicans hyphal formation [30,31].
These effects, in addition to the generation of a hyphae-inhibiting, acidic environment,
can explain why yeast forms are associated with S. mutans clusters in the deeper layers
of mixed biofilms [12]. Furthermore, competence-stimulating peptides released by S.
mutans [32] and S. gordonii [33] also disrupt hyphal formation in C. albicans cells.
These hyphae-inhibiting effects are consistent with the fact that the Efg1 filamentation
pathway is not required for mixed C. albicans–S. mutans biofilm growth and cariogenicity
as noted above, suggesting that in contrast to mucosal candidiasis, filamentation
may not be a virulence-promoting phenotype in mineralized tissue infections such as
dental caries. Paradoxically, farnesol produced by C. albicans, which stimulates S.
mutans growth and gtfB expression at low concentrations (25–50 μM), disrupts bacterial
growth at high concentrations (>100 μM) [27]. Therefore, a tightly regulated cooperative
and antagonistic balance through stimulus-inhibition mechanisms appears to mediate
bacterial–fungal coexistence and survival within biofilms, which can become synergistic
when conditions are conducive for disease (Fig 2).
In summary, the polymicrobial nature of biofilm-associated oral diseases has been
increasingly recognized. Clinical data, together with in vivo studies, provide compelling
evidence of the importance of cross-kingdom interactions in the severity of mucosal
diseases and dental caries. Complex cell–cell and cell–EPS matrix interactions, spatial
organization, and chemical/metabolic factors modulate biofilm development and virulence.
These fungal–bacterial interactions are facilitated by host factors (immunity, diet,
and salivary function) to modify the local microenvironment and promote oral diseases.
Elucidating how bacterial–fungal interactions occur spatiotemporally (cooperative,
competitive, or both simultaneously) to mediate symbiotic, antagonistic, or synergistic
states may shed new light into the pathogenic mechanisms and identify more effective
therapeutic targets. Since cross-kingdom biofilms exist throughout the gastrointestinal
tract, principles and molecules that emerge from these studies may lead to novel approaches
to prevent and eradicate other intractable polymicrobial biofilms at various clinical
niches.
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
Symbiotic relationship between Streptococcus mutans and Candida albicans synergizes virulence of plaque biofilms in vivo.
Marlise Klein, Gene Watson, Megan Falsetta … (2014)
- Record: found
- Abstract: found
- Article: not found
Dental biofilm: ecological interactions in health and disease
P D Marsh, Egija Zaura (2017)