- Record: found
- Abstract: found
- Article: found
The Teratogenic Effects of Prenatal Ethanol Exposure Are Exacerbated by Sonic Hedgehog or Gli2 Haploinsufficiency in the Mouse
Read this article at
Abstract
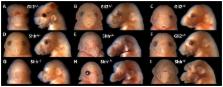
Disruption of the Hedgehog signaling pathway has been implicated as an important molecular mechanism in the pathogenesis of fetal alcohol syndrome. In severe cases, the abnormalities of the face and brain that result from prenatal ethanol exposure fall within the spectrum of holoprosencephaly. Single allele mutations in the Hh pathway genes Sonic Hedgehog ( SHH) and GLI2 cause holoprosencephaly with extremely variable phenotypic penetrance in humans. Here, we tested whether mutations in these genes alter the frequency or severity of ethanol-induced dysmorphology in a mouse model. Timed pregnancies were established by mating Shh +/− or Gli2 +/− male mice backcrossed to C57BL/6J strain, with wildtype females. On gestational day 7, dams were treated with two ip doses of 2.9 g/kg ethanol (or vehicle alone), administered four hrs apart. Fetuses were then genotyped and imaged, and the severity of facial dysmorphology was assessed. Following ethanol exposure, mean dysmorphology scores were increased by 3.2- and 6.6-fold in Shh +/− and Gli2 +/− groups, respectively, relative to their wildtype littermates. Importantly, a cohort of heterozygous fetuses exhibited phenotypes not typically produced in this model but associated with severe holoprosencephaly, including exencephaly, median cleft lip, otocephaly, and proboscis. As expected, a correlation between the severity of facial dysmorphology and medial forebrain deficiency was observed in affected animals. While Shh +/− and Gli2 +/− mice have been described as phenotypically normal, these results illustrate a functional haploinsufficiency of both genes in combination with ethanol exposure. By demonstrating an interaction between specific genetic and environmental risk factors, this study provides important insights into the multifactorial etiology and complex pathogenesis of fetal alcohol syndrome and holoprosencephaly.
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
Fetal alcohol syndrome: embryogenesis in a mouse model.
- Record: found
- Abstract: found
- Article: not found
Molecular interactions coordinating the development of the forebrain and face.
- Record: found
- Abstract: found
- Article: not found