- Record: found
- Abstract: found
- Article: found
Comprehensive Analysis of Quantitative Proteomics With DIA Mass Spectrometry and ceRNA Network in Intrahepatic Cholestasis of Pregnancy
Read this article at
Abstract
Background: Intrahepatic cholestasis of pregnancy (ICP) is a pregnancy-specific complication characterized by pruritus without skin damage and jaundice. The poor perinatal outcomes include fetal distress, preterm birth, and unexpected intrauterine death. However, the mechanism of ICP leading to poor prognosis is still unclear.
Methods: We analyzed 10 ICP and 10 normal placental specimens through quantitative proteomics of data-independent acquisition (DIA) to screen and identify differentially expressed proteins. GO, KEGG, COG/KOG, StringDB, InterProScan, Metascape, BioGPS, and NetworkAnalyst databases were used in this study. PITA, miRanda, TargetScan, starBase, and LncBase Predicted v.2 were used for constructing a competing endogenous RNA (ceRNA) network. Cytoscape was used for drawing regulatory networks, and cytoHubba was used for screening core nodes. The ICP rat models were used to validate the pathological mechanism.
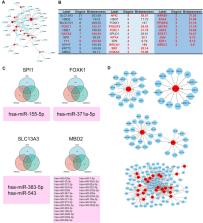
Results: GO, KEGG, and COG/KOG functional enrichment analysis results showed the differentially expressed proteins participated in autophagy, autophagosome formation, cofactor binding, JAK-STAT signaling pathway, and coenzyme transport and metabolism. DisGeNET analysis showed that these differentially expressed proteins were associated with red blood cell disorder and slow progression. We further analyzed first 12 proteins in the upregulated and downregulated differentially expressed proteins and incorporated clinicopathologic parameters. Our results showed HBG1, SPI1, HBG2, HBE1, FOXK1, KRT72, SLC13A3, MBD2, SP9, GPLD1, MYH7, and BLOC1S1 were associated with ICP development. ceRNA network analysis showed that MBD2, SPI1, FOXK1, and SLC13A3 were regulated by multiple miRNAs and lncRNAs.
Conclusion: ICP was associated with autophagy. The ceRNA network of MBD2, SPI1, FOXK1, and SLC13A3 was involved in ICP progression, and these core proteins might be potential target.
Related collections
Most cited references37
- Record: found
- Abstract: found
- Article: not found
Cytoscape: a software environment for integrated models of biomolecular interaction networks.

- Record: found
- Abstract: found
- Article: found
The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets

- Record: found
- Abstract: found
- Article: found