- Record: found
- Abstract: found
- Article: found
Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways
Read this article at
Abstract
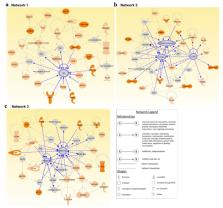
Evidence of familial inheritance in non-medullary thyroid cancer (NMTC) has accumulated over the last few decades. However, known variants account for a very small percentage of the genetic burden. Here, we focused on the identification of common pathways and networks enriched in NMTC families to better understand its pathogenesis with the final aim of identifying one novel high/moderate-penetrance germline predisposition variant segregating with the disease in each studied family. We performed whole genome sequencing on 23 affected and 3 unaffected family members from five NMTC-prone families and prioritized the identified variants using our Familial Cancer Variant Prioritization Pipeline (FCVPPv2). In total, 31 coding variants and 39 variants located in upstream, downstream, 5′ or 3′ untranslated regions passed FCVPPv2 filtering. Altogether, 210 genes affected by variants that passed the first three steps of the FCVPPv2 were analyzed using Ingenuity Pathway Analysis software. These genes were enriched in tumorigenic signaling pathways mediated by receptor tyrosine kinases and G-protein coupled receptors, implicating a central role of PI3K/AKT and MAPK/ERK signaling in familial NMTC. Our approach can facilitate the identification and functional validation of causal variants in each family as well as the screening and genetic counseling of other individuals at risk of developing NMTC.
Related collections
Most cited references36
- Record: found
- Abstract: found
- Article: not found
dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs.
- Record: found
- Abstract: found
- Article: not found
Germline mutations in the proof-reading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas
- Record: found
- Abstract: found
- Article: not found