- Record: found
- Abstract: found
- Article: not found
Timing, rates and spectra of human germline mutation
Read this article at

Abstract
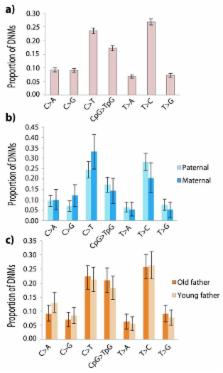
Germline mutations are a driving force behind genome evolution and genetic disease. We investigated genome-wide mutation rates and spectra in multi-sibling families. Mutation rate increased with paternal age in all families, but the number of additional mutations per year differed more than two-fold between families. Meta-analysis of 6,570 mutations showed that germline methylation influences mutation rates. In contrast to somatic mutations, we found remarkable consistency of germline mutation spectra between the sexes and at different paternal ages. 3.8% of mutations were mosaic in the parental germline, resulting in 1.3% of mutations being shared between siblings. The number of these shared mutations varied significantly between families. Our data suggest that the mutation rate per cell division is higher during both early embryogenesis and differentiation of primordial germ cells, but is reduced substantially during post-pubertal spermatogenesis. These findings have important consequences for the recurrence risks of disorders caused by de novo mutations.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
Analysis of genetic inheritance in a family quartet by whole-genome sequencing.
- Record: found
- Abstract: found
- Article: not found
Toward better understanding of artifacts in variant calling from high-coverage samples.
- Record: found
- Abstract: found
- Article: not found