- Record: found
- Abstract: found
- Article: found
Cisplatin ototoxicity mechanism and antagonistic intervention strategy: a scope review
Read this article at
Abstract
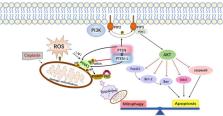
Cisplatin is a first-line chemotherapeutic agent in the treatment of malignant tumors with remarkable clinical effects and low cost. However, the ototoxicity and neurotoxicity of cisplatin greatly limit its clinical application. This article reviews the possible pathways and molecular mechanisms of cisplatin trafficking from peripheral blood into the inner ear, the toxic response of cisplatin to inner ear cells, as well as the cascade reactions leading to cell death. Moreover, this article highlights the latest research progress in cisplatin resistance mechanism and cisplatin ototoxicity. Two effective protective mechanisms, anti-apoptosis and mitophagy activation, and their interaction in the inner ear are discussed. Additionally, the current clinical preventive measures and novel therapeutic agents for cisplatin ototoxicity are described. Finally, this article also forecasts the prospect of possible drug targets for mitigating cisplatin-induced ototoxicity. These include the use of antioxidants, inhibitors of transporter proteins, inhibitors of cellular pathways, combination drug delivery methods, and other mechanisms that have shown promise in preclinical studies. Further research is needed to evaluate the efficacy and safety of these approaches.
Related collections
Most cited references89
- Record: found
- Abstract: found
- Article: not found
Ubiquitin is phosphorylated by PINK1 to activate parkin.

- Record: found
- Abstract: found
- Article: found
Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions

- Record: found
- Abstract: found
- Article: found