- Record: found
- Abstract: found
- Article: found
A STAT3 inhibitor ameliorates CNS autoimmunity by restoring Teff:Treg balance

Read this article at
Abstract
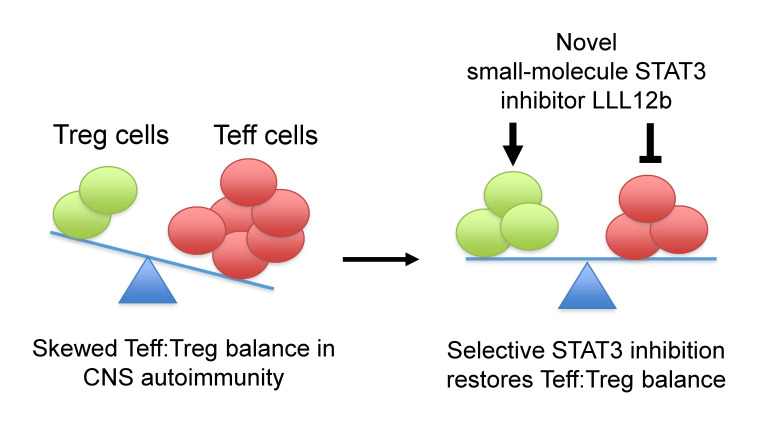
Reestablishing an appropriate balance between T effector cells (Teff) and Tregs is essential for correcting autoimmunity. Multiple sclerosis (MS) is an immune-mediated chronic CNS disease characterized by neuroinflammation, demyelination, and neuronal degeneration, in which the Teff:Treg balance is skewed toward pathogenic Teffs Th1 and Th17 cells. STAT3 is a key regulator of Teff:Treg balance. Using the structure-based design, we have developed a potentially novel small-molecule prodrug LLL12b that specifically inhibits STAT3 and suppresses Th17 differentiation and expansion. Moreover, LLL12b regulates the fate decision between Th17 and Tregs in an inflammatory environment, shifting Th17:Treg balance toward Tregs and favoring the resolution of inflammation. Therapeutic administration of LLL12b after disease onset significantly suppresses disease progression in adoptively transferred, chronic, and relapsing-remitting experimental autoimmune encephalomyelitis. Disease relapses were also significantly suppressed by LLL12b given during the remission phase. Additionally, LLL12b shifts Th17:Treg balance of CD4 + T cells from MS patients toward Tregs and increases Teff sensitivity to Treg-mediated suppression. These data suggest that selective inhibition of STAT3 by the small molecule LLL12b recalibrates the effector and regulatory arms of CD4 + T responses, representing a potentially clinically translatable therapeutic strategy for MS.
Abstract
Related collections
Most cited references76
- Record: found
- Abstract: found
- Article: not found
Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain.
- Record: found
- Abstract: found
- Article: not found
Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells.
- Record: found
- Abstract: found
- Article: not found