- Record: found
- Abstract: found
- Article: found
Electrophysiological Abnormalities in Angelman Syndrome Correlate With Symptom Severity
Read this article at
Abstract
BACKGROUND:
Angelman syndrome (AS) is a rare neurodevelopmental disorder caused by the absence of functional UBE3A in neurons. Excess low-frequency oscillations as measured with electroencephalography (EEG) have been identified as a characteristic finding, but the relationship of this EEG finding to the symptomatology of AS and its significance in the pathophysiology of AS remain unknown.
METHODS:
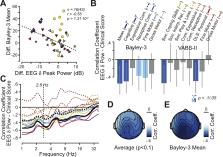
We used correlations and machine learning to investigate the cross-sectional and longitudinal relationship between EEG spectral power and motor, cognitive, and language skills (Bayley Scales of Infant and Toddler Development, Third Edition); adaptive behavior (Vineland Adaptive Behavior Scales, Second Edition); AS-specific symptoms (AS Clinical Severity Scale); and the age of epilepsy onset in a large sample of children (age: 1–18 years) with AS due to a chromosomal deletion of 15q11-q13 (45 individuals with 72 visits).
RESULTS:
We found that after accounting for age differences, participants with stronger EEG delta-band abnormality had earlier onset of epilepsy and lower performance scores across symptom domains including cognitive, motor, and communication. Combing spatial and spectral information beyond the delta frequency band increased the cross-sectional association with clinical severity on average by approximately 45%. Furthermore, we found evidence for longitudinal correlations of EEG delta-band power within several performance domains, including the mean across Bayley Scales of Infant and Toddler Development, Third Edition, scores.
CONCLUSIONS:
Our results show an association between EEG abnormalities and symptom severity in AS, underlining the significance of the former in the pathophysiology of AS. Furthermore, our work strengthens the rationale for using EEG as a biomarker in the development of treatments for AS, a concept that may apply more generally to neurodevelopmental disorders.
Related collections
Most cited references25
- Record: found
- Abstract: not found
- Article: not found
Comparing correlated correlation coefficients.
- Record: found
- Abstract: not found
- Article: not found
The Proof and Measurement of Association between Two Things
- Record: found
- Abstract: found
- Article: not found