- Record: found
- Abstract: found
- Article: not found
A Probabilistic Model of Local Sequence Alignment That Simplifies Statistical Significance Estimation
Read this article at

Abstract
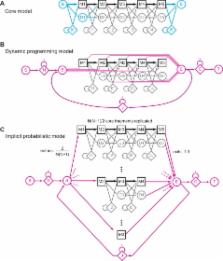
Sequence database searches require accurate estimation of the statistical significance of scores. Optimal local sequence alignment scores follow Gumbel distributions, but determining an important parameter of the distribution ( λ) requires time-consuming computational simulation. Moreover, optimal alignment scores are less powerful than probabilistic scores that integrate over alignment uncertainty (“Forward” scores), but the expected distribution of Forward scores remains unknown. Here, I conjecture that both expected score distributions have simple, predictable forms when full probabilistic modeling methods are used. For a probabilistic model of local sequence alignment, optimal alignment bit scores (“Viterbi” scores) are Gumbel-distributed with constant λ = log 2, and the high scoring tail of Forward scores is exponential with the same constant λ. Simulation studies support these conjectures over a wide range of profile/sequence comparisons, using 9,318 profile-hidden Markov models from the Pfam database. This enables efficient and accurate determination of expectation values (E-values) for both Viterbi and Forward scores for probabilistic local alignments.
Author Summary
Sequence database searches are a fundamental tool of molecular biology, enabling researchers to identify related sequences in other organisms, which often provides invaluable clues to the function and evolutionary history of genes. The power of database searches to detect more and more remote evolutionary relationships – essentially, to look back deeper in time – has improved steadily, with the adoption of more complex and realistic models. However, database searches require not just a realistic scoring model, but also the ability to distinguish good scores from bad ones – the ability to calculate the statistical significance of scores. For many models and scoring schemes, accurate statistical significance calculations have either involved expensive computational simulations, or not been feasible at all. Here, I introduce a probabilistic model of local sequence alignment that has readily predictable score statistics for position-specific profile scoring systems, and not just for traditional optimal alignment scores, but also for more powerful log-likelihood ratio scores derived in a full probabilistic inference framework. These results remove one of the main obstacles that have impeded the use of more powerful and biologically realistic statistical inference methods in sequence homology searches.
Related collections
Most cited references51
- Record: found
- Abstract: not found
- Article: not found
Identification of common molecular subsequences.
- Record: found
- Abstract: found
- Article: not found