- Record: found
- Abstract: found
- Article: not found
Mitochondrial DNA depletion and fatal infantile hepatic failure due to mutations in the mitochondrial polymerase γ (POLG) gene: a combined morphological/enzyme histochemical and immunocytochemical/biochemical and molecular genetic study
Read this article at

Abstract
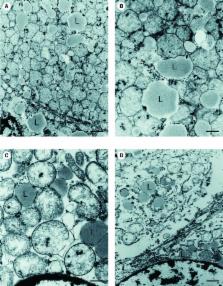
Combined morphological, immunocytochemical, biochemical and molecular genetic studies were performed on skeletal muscle, heart muscle and liver tissue of a 16-months boy with fatal liver failure. The pathological characterization of the tissues revealed a severe depletion of mtDNA (mitochondrial DNA) that was most pronounced in liver, followed by a less severe, but still significant depletion in skeletal muscle and the heart. The primary cause of the disease was linked to compound heterozygous mutations in the polymerase γ (POLG) gene (DNA polymerase γ; A467T, K1191N). We present evidence, that compound heterozygous POLG mutations lead to tissue selective impairment of mtDNA replication and thus to a mosaic defect pattern even in the severely affected liver. A variable defect pattern was found in liver, muscle and heart tissue as revealed by biochemical, cytochemical, immunocytochemical and in situ hybridization analysis. Functionally, a severe deficiency of cytochrome-c-oxidase (cox) activity was seen in the liver. Although mtDNA depletion was detected in heart and skeletal muscle, there was no cox deficiency in these tissues. Depletion of mtDNA and microdissection of cox-positive or negative areas correlated with the histological pattern in the liver. Interestingly, the mosaic pattern detected for cox-activity and mtDNA copy number fully aligned with the immunohistologically revealed defect pattern using Pol γ, mtSSB- and mtTFA-antibodies, thus substantiating the hypothesis that nuclear encoded proteins located within mitochondria become unstable and are degraded when they are not actively bound to mtDNA. Their disappearance could also aggravate the mtDNA depletion and contribute to the non-homogenous defect pattern.
Related collections
Most cited references52
- Record: found
- Abstract: found
- Article: not found
Mitochondrial DNA maintenance in vertebrates.
- Record: found
- Abstract: found
- Article: not found
Mitochondrial disorders.
- Record: found
- Abstract: not found
- Article: not found