- Record: found
- Abstract: found
- Article: not found
Integrated Genomic Analyses of Ovarian Carcinoma
Read this article at

Summary
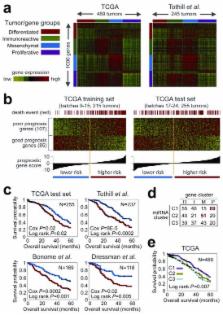
The Cancer Genome Atlas (TCGA) project has analyzed mRNA expression, miRNA expression, promoter methylation, and DNA copy number in 489 high-grade serous ovarian adenocarcinomas (HGS-OvCa) and the DNA sequences of exons from coding genes in 316 of these tumors. These results show that HGS-OvCa is characterized by TP53 mutations in almost all tumors (96%); low prevalence but statistically recurrent somatic mutations in 9 additional genes including NF1, BRCA1, BRCA2, RB1, and CDK12; 113 significant focal DNA copy number aberrations; and promoter methylation events involving 168 genes. Analyses delineated four ovarian cancer transcriptional subtypes, three miRNA subtypes, four promoter methylation subtypes, a transcriptional signature associated with survival duration and shed new light on the impact on survival of tumors with BRCA1/2 and CCNE1 aberrations. Pathway analyses suggested that homologous recombination is defective in about half of tumors, and that Notch and FOXM1 signaling are involved in serous ovarian cancer pathophysiology.
Related collections
Most cited references34
- Record: found
- Abstract: found
- Article: not found
The emerging roles of forkhead box (Fox) proteins in cancer.
- Record: found
- Abstract: found
- Article: not found
Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma.

- Record: found
- Abstract: found
- Article: found