- Record: found
- Abstract: found
- Article: found
The Mechanism of Markovnikov-Selective Epoxide Hydrogenolysis Catalyzed by Ruthenium PNN and PNP Pincer Complexes
Read this article at
Abstract

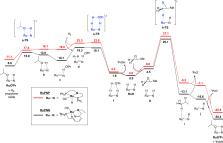
The homogeneous catalysis of epoxide hydrogenolysis to give alcohols has recently received significant attention. Catalyst systems have been developed for the selective formation of either the Markovnikov (branched) or anti-Markovnikov (linear) alcohol product. Thus far, the reported catalysts exhibiting Markovnikov selectivity all feature the potential for Noyori/Shvo-type bifunctional catalysis, with either a RuH/NH or FeH/OH core structure. The proposed mechanisms of epoxide ring-opening have involved cooperative C–O bond hydrogenolysis involving the metal hydride and the acidic pendant group on the ligand, in analogy to the well-documented mechanism of polar double-bond hydrogenation exhibited by catalysts of this type. In this work, we present a combined computational/experimental study of the mechanism of epoxide hydrogenolysis catalyzed by Noyori-type PNP and PNN complexes of ruthenium. We find that, at least for these ruthenium systems, the previously proposed bifunctional pathway for epoxide ring-opening is energetically inaccessible; instead, the ring-opening proceeds through opposite-side nucleophilic attack of the ruthenium hydride on the epoxide carbon, without the involvement of the ligand N–H group. For both catalyst systems, the rate law and overall barrier predicted by density functional theory (DFT) are consistent with the results from kinetic studies.
Related collections
Most cited references64
- Record: found
- Abstract: not found
- Article: not found
Mercury: visualization and analysis of crystal structures
- Record: found
- Abstract: found
- Article: not found
How to conceptualize catalytic cycles? The energetic span model.
- Record: found
- Abstract: not found
- Article: not found